To demonstrate that LZC696 (L) reduces organ damage in an experimental model of metabolic syndrome, were explored two mechanisms: anti-inflammatory effects through the IL-6Ralpha pathway and through MAS1R, the production of endothelial repair mediated by VEGFR2+/CD133+ endothelial progenitor cells (EPCs). Experimental model of metabolic syndrome was realized by WKY rats and SHRs. SHR and WKY received a fructose diet in drinking water at 10% v/v for 12 weeks (FFHR and FFR receptivity). Chronic treatment with L: (68 mg / kg per day for 6 weeks) and valsartan (V) (34 mg / kg per day for 6 weeks, as control equimolar group. Was determined: SBP, fast glycaemia and TTGO, left ventricular hypertrophy (HVI), vascular remodelling, hsCPR expression, and vascular expression in mesenteric tissue of IL-6Ralfa, STAT3, VEGFR2 and CD133 were determined. The experimental model was confirmed. L treatment reverted SBP, HVI, remodelling and vascular inflammation, decreased STAT3 expression and hsCPR in FFHR. Additionally, the most important finding was that L produced an increase in the expression of resident EPCs in the endothelial tissue of mesenteric tissue.
LZC696, metabolic syndrome, IL6R-alpha, vascular inflammation, vascular remodelling, hypertension
Cardiovascular disease is the most important cause of death in the world. Hypertension, coronary heart disease and heart failure are the manifestations of a very advanced vascular disease.
LCZ696 is the first of a new class of drugs that simultaneously block ARB (Angiotensin 1 receptor blocker) and NEP (neprylisin or neutral endopeptidase protein); hence, they are referred to with the acronym ARNi [1-3].
This complex system results in multiple effects on the cardiovascular system. In the first instance, LCZ696 can increase the half-life of BNP through the initiation of NEP, managing to increase natriuresis and vasodilation through activation of the NPRA receptor. On the other hand, the blockade of AT1R can decrease fibrosis, induce vasodilation, reduce the retention of sodium and water, lower blood pressure and other effects [4].
The most important clinical study that demonstrated the reduction of cardiovascular morbidity and mortality in patients with heart failure was the PARADIGM-HF study, which showed a clear benefit in patients who were in the branch receiving LCZ696. However, studies in experimental animals and pathophysiological analyses are scarce [3].
Thus, the precise mechanism by which LCZ696 reduces cardiovascular mortality remains unclear. Some authors have proposed different hypotheses: 1- a sustained increase in natriuretic peptides by inhibition of NEP; 2- a direct hemodynamic effect that reduces stress on the left ventricular wall; 3- a reduction in arrhythmias by a reduction in fibrosis or myocardial hypertrophy; 4- an improvement of regional myocardial perfusion [4].
To investigate these hypotheses, the experimental model proposed in this paper consists of rats spontaneously ingesting a diet rich in the combination of carbohydrates and fructose, which in previous studies has been shown to produce significant remodelling and vascular inflammation. In addition, this model is useful to evaluate the epigenetic effects of fructose in rats genetically predisposed to developing arterial hypertension [5-7].
Previously, drug-intervention studies in this model have shown that some anti-hypertensive treatments, with candesartan, telmisartan and losartan, not only lower blood pressure but also cause an improvement in redox balance and regression of structural changes in resistance arteries, although not regeneration of the endothelium. In other words, reversing adverse effects associated with hypertension does not improve the normal structure and function of endothelial cells [8,9].
Within the inflammatory cascades activated in this experimental model, we found that the determinant of the previously evidenced changes is that of the IL-6 receptor. Two different forms of IL-6 cell receptors have been described: an 80 kDa ligand binding chain, known as IL-6R (IL-6Ra, CD126), and a 130 kDa signal transduction chain, gp130 (IL - 6Rb, CD130). Gp130 is present in many places and situations; in contrast, IL-6R shows a more limited expression pattern [10].
The activation cascade begins with the binding of IL-6 to IL-6R in target cells, and then the complex of IL-6 and IL-6R encounters gp130, thus increasing its dimerization and subsequent activation of intracellular signalling, such as the phosphorylation of STAT3 by JAK. This so-called classical signalling pathway activates the expression of several acute phase proteins, such as C-reactive protein. STAT3 resides in the cytoplasm. The activated STAT3 complex will translocate from the cytoplasm to the nucleus, which initiates the transcription of STAT3 target genes [11,12].
Repair of the denuded endothelium might be crucial for the restoration of endothelial function. Endothelial progenitor cells (EPCs) have been regarded as restorative cells that home to sites of endothelial damage and denudation. Furthermore, the activities of EPCs have been shown to correlate with the functional integrity of the endothelium [13,14].
The definitions of EPC are nonetheless varied and controversial, which results in the generation of a complicated list of putative EPC immunophenotypes [13-15]. Although recent data suggest that CD133+CD34+VEGF receptor 2 (VEGFR2) + cells do not appear to be true EPCs, CD133+, CD34+, or VEGFR2+, or any combination of these immunophenotypes, have been commonly used to enumerate putative EPCs [13-15]. There is evidence that both circulating and resident EPCs at the vascular level are markers of cardiovascular risk and subclinical atherogenesis [15-17].
The aim of the work was to demonstrate the protective effect of dual NEP/ARB inhibition at the vascular level. To demonstrate this, we explored two mechanisms: anti-inflammatory effects through the IL-6Ralpha pathway and through MAS1R, the production of endothelial repair mediated by VEGFR2+/CD133+ endothelial progenitor cells (EPCs).
Experimental design
Wistar Kyoto (WKY) and spontaneously hypertensive rats (SHR), all males, were separated into 4 groups: 1-WKY: Control, 2-FFR: WKY rats receiving a 10% (w/v) fructose solution for 12 weeks, 3-SHR: Control, 4-FFHR: SHR receiving a 10% fructose solution (p/v) for 12 weeks; After 12 weeks, the rats were randomly divided into 5-FFHR+L: (68 mg/kg per day for 6 weeks) and 6-FFHR+V: (34 mg/kg per day for 6 weeks) (n = 6 each group), plus corresponding control groups.
These animals were granted by the Bioterium of School of Medicine of the National University of Cuyo after the approval of the Institutional Committee for the Care and Use of Laboratory Animals (CICUAL). CICAL resolution No. 91/2016.
After an overnight fast, rats were weighed and anesthetized with ketamine (50 mg/kg) and acepromazine (1 mg/kg). Blood was collected from the abdominal aorta into heparinized tubes. Plasma was obtained after centrifugation at 1,000 x g for 15 min at 4°C. Epididymal adipose tissue was collected, weighed, and stored frozen at -80°C until assayed.
Systolic blood pressure measurement
During the different stages of the protocol, systolic blood pressure (SBP) determinations were made by the plethysmography method in the tail artery using the CODA2 system (Kent Scientific, USA).
Biochemical determinations
Through blood samples obtained from the tail of the animal, the following determinations were generated: lipid profile (total cholesterol, HDL, triglycerides), highly sensitive CRP, fasting and postprandial glycaemia. The extractions were performed at the beginning of the experimental drugs and at the end of the protocol.
At 18 weeks of the animals’ life, and after meeting the stipulated time of administration of the drug in question, we proceeded to sacrifice by the exsanguination method following the international recommendations for the use of experimental animals.
The organs obtained were processed for different uses, including inclusion in paraffin and freezing at -80 °C.
Histomorphometry
The heart weights of the different groups were compared in the analysis with the total body weight and the muzzle length in each of the animals. The sections of tissues included in paraffin were stained with Masson's Trichrome technique and the degree of hypertrophy of myocardiocytes was observed with a NIKON 80i microscope. Similarly, the mesenteric and/or coronary arteries were processed to estimate the lumen/mean (L/M) ratio using digital microphotography, and images were analysed with Image J software.
Western Blot (WB) technique
The mesenteric tissue from each rat was washed in PBS, and the proteins were extracted in cold 20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X-100, and a protease inhibitor mixture (P2714, Sigma). After sonication for 15 s (3 times with 10-s intervals) and extraction for 30 min at 4°C, the sample extracts were clarified by centrifugation at 14,000 x g for 20 min and used immediately or stored at -20°C. The proteins were separated on 10% polyacrylamide slab gels and transferred to 0.22-µm nitrocellulose membranes (GE, Germany). Nonspecific reactivity was blocked by incubation for 1h at room temperature in 5% non-fat dry milk dissolved in washing buffer (PBS, pH 7.6, 0.2% Tween 20). The expression of different proteins in the homogenates of mesenteric arteries was evaluated using the following antibodies: Anti-Vascular Endothelial Growth Factor Receptor-2 (VEGF R2) (Sigma Aldrich V1014), Anti-CD133 (Prominin-1, Sigma Aldrich C9493), Anti-STAT 3 (Enzo 7G8 ADI-905-756-100), Anti-Receptor Interleukin 6 alpha (IL-6Rα) (Santa Cruz Biotechnology (D-8): sc-374259) and Anti-MAS1 Santa Cruz Biotechnology (G-1): sc-390453). Excess first and second antibodies were removed by washing 5 times for 5 min in blocking solution. Detection was accomplished with an enhanced chemiluminescence system (ABC, Dako System) and subsequent exposure to Kodak X-AR film (Eastman Kodak) for 5-30s.
Immunohistochemistry
Anti-vascular endothelial growth factor receptor-2 (VEGFR2) was used for immunohistochemistry. Tissue sections were cut at a 3-μm thickness from paraffin-embedded blocks. The antibodies were diluted 1:1000. The primary incubations were carried out for 1 hour at 21-22°C, followed by six extensive washes in PBS with Triton X-100 for 5 min each. The secondary antibodies, anti-rabbit HRP and anti-goat IgG HRP (Sigma-Aldrich), were diluted in PBS alone according to the manufacturer's instructions.
The images were collected with Nikon EZ-C1 3.00 software on a Nikon Diaphot TMD microscope.
Reagents
Unless otherwise noted, all reagents were purchased from Sigma Chemical Co, MO USA.
Statistical and data analysis
The data are expressed as the mean ± SEM. The statistical significance of the comparisons between all groups was assessed by one-way ANOVA followed by the Bonferroni post-test. A two-sided p value of less than 0.05 was considered significant.
Initially, variables related to the confirmation of the experimental model were analysed; see table 1. FFHR showed, with respect to other groups, significant increases in systolic blood pressure, glycaemia, area under the curve in oral glucose tolerance test (GTT), and triglycerides, with a significant decrease in HDL cholesterol. These results confirm the experimental model. Regarding the treatment with L, no significant changes were observed in the metabolic variables studied, while there was a significant difference in SBP (Table 1). With the treatment with V, a similar pattern occurred, and there were no significant differences in the metabolic variables, but there was a significant difference in SBP. There were only significant differences between the groups with treatment in the SBP values , showing a more important decrease in the group treated with L with respect to V.
Table 1. Metabolic and cardiovascular variables
Variable |
WKY |
SHR |
FHHR |
FFHR+L |
FFHR+V |
Fasting glucose (mmol/L) |
4.2±0.03 |
4.8.0±0.07 |
6.2±0.05* |
6.1±0.1* |
6.4±0.09* |
Fasting triglycerides (mmol/L) |
0.68±0.1 |
0.95±0.03 |
1.5±0.02* |
1.3±0.06* |
1.5±0.12* |
HOMA (µU/mL insulin x mmol/L glucose) / 22.5 |
5.2±0.08 |
6.1±0.05* |
13±0.06* |
13.2±0.03* |
11.1±0.04* |
HDL Cholesterol (mmol/L) |
0.5±0.04 |
0.4±0.0 |
0.28±0.01* |
0.3±0.01* |
0.3±0.03* |
High-sensitivity C Reactive Protein (mg/dL) |
1.09±0.01 |
3.1±0.03 |
5.4±0.01* |
0.6±0.01*# |
4.2±0.29* |
Systolic Blood Pressure (mmHg) |
130.8±4.9 |
155±4.1 |
171±5.3 |
128.3±1.2 |
147.5±2.7 |
Metabolic and Hemodynamic variables. Mean ± SEM. * > p.001 v WKY and # > p.001 v FFHR
Then, we analysed the impact on the myocardium as a target organ damage (OD) of this syndrome. Different techniques were used that allowed us to adequately corroborate left ventricular hypertrophy (LVH), and in turn, this is adequate and reproducible. The pathological experimental model, as well as SHR, clearly developed LVH. This demonstrates the impact of the metabolic and hemodynamic changes that this experimental model (FFHR) had. The weight-cardiac / body-weight ratio, the weight-cardiac / snout-anus length ratio and measurement of the myocardiocyte area in the left ventricular free wall showed significant differences in FFHR and SHR (Figure 1).
Treatment with L reversed LVH in the 3 assessment methods, showing significant differences with respect to FFHR, and in the case of the myocardiocyte area assessment, there was a significant decrease with respect to V. Treatment with V showed significant differences with respect to FFHR in the 3 methods studied (Figure 1).
For the analysis of vascular remodelling from the determination of the lumen / media relation, the FFHR model, with respect to the control, WKY and SHR, showed significant differences showing vascular damage. Chronic treatment with L restored the vascular structure to the values of the control group, in addition a significant increase with respect to V. V was able to revert the vascular changes, in a significant manner with respect to FFHR (Figure 1).
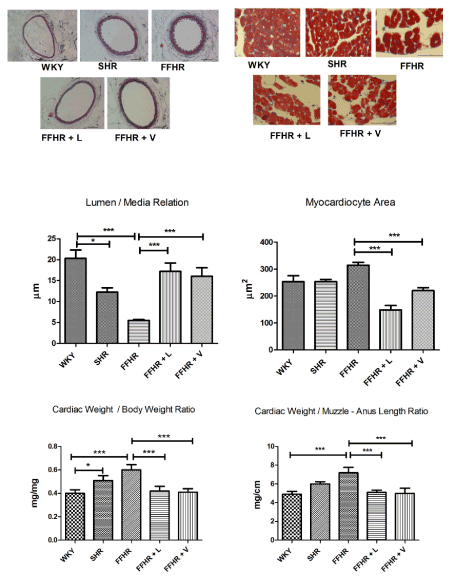
Figure 1. Analysed the impact on the myocardium and vascular remodelling as a target organ damage (OD) of this syndrome. In the upper part, you can see representative images of, to the right, carotid arteries (200x), and to the left, free wall of the left ventricle (x1000). At the bottom of the graphic presentation of statistical analysis is observed. Different techniques were used that allowed us to adequately corroborate left ventricular hypertrophy (LVH). Masson's trichromic stain. Mean ± SD. * > p.001 v WKY and # > p.001 v FFHR
The results of the inflammatory variables showed that the experimental model showed a significant increase in hsCPR with respect to the control group.
To evaluate this inflammatory pathway, expression of the IL-6 receptor alpha subunit (IL6Ra) was studied. FFHR did not show significant differences in expression with respect to the control group, but the expression of STAT3 showed hyperactivity of this intracellular pathway. The treatment with L significantly increased the expression of IL-6Ra with respect to FFHR as well as V but reduced the activation of STAT3 through a reduction in expression after treatment with L and V (Figure 2).
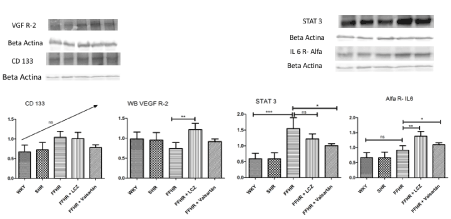
Figure 2. Western Blot representative images. In the lower part of each graphic representation we can see the image of the development of the WB of each protein with its respective load control. In this image, the expression of IL-6R alpha, STAT3, VEGFR2, CD133 and MAS1R is shown. Mean ± SD. * > p.001 v WKY and # > p.001 v FFHR
Additionally, vascular repair was evaluated based on the expression of cell membrane markers and cluster differentiation (CD). Mature endothelial progenitor cells, including VEGFR2 and CD133, were evaluated. The experimental model showed a significant reduction in the expression of both markers, which correlates with a lower presence at the vascular level, in the mesenteric arteries. Chronic treatment with L reverses these changes with respect to FFHR, showed similar expression to WKY group. Treatment with V did not show significant differences with respect to the pathological experimental model (Figure 2).
In addition, the expression of MAS1 was studied in the experimental model. WB analysis showed that MAS expression was significantly lower in FFHR versus WKY. After treatment with L, the expression of MAS was significantly higher than in FFHR and that after treatment with V. V increased the expression of MAS with respect to FFHR (Figure 2).
The most important finding of this study was to have found important changes at the vascular level, from the treatment with LCZ696. The reversal of vascular changes associated with the metabolic syndrome, both in the lumen / mean ratio and at the cardiac muscle level, could explain the results found in the randomized clinical trials.
The experimental model, as demonstrated in previous publications, was corroborated. FFHR group, in addition to presenting alterations in metabolic variables, showed a very significant vascular inflammatory process [8]. This model presented metabolic disorders as dyslipidaemia and insulin resistance, and hypertension. Treatment with LCZ696 was not able to modify the metabolic variables, similarly to valsartan alone. These results support the results that valsatan does not have an insulin-sensitizing effect and that dual inhibition does not provide major changes with respect to the latter [18,19].
Regarding the effect on SBP, both treatments significantly reduced these values, with LCZ696 being more effective in this reduction, showing significant differences with respect to valsartan. This supports the hypothesis that LCZ696 has more important hemodynamic effects, including a greater reduction in intravascular volume, which probably leads to an equal dose of valsatan, a greater reduction in SBP, by increasing the half-life of natriuretic peptides [20,21].
Regarding the protective effect on white organ damage of hypertension were evaluated LVH and vascular remodelling. FFHR and SHR developed LVH. FFHR presented greater LVH. This result has already been discussed in previous publications. This effect was probably mediated by not only a hemodynamic stimulus, but also by metabolic changes in the activation of growth factors. Treatment with LCZ696 reverted these changes, and when the histology structure of the LV wall was analysed, showing an even more significant reduction with LCZ696 respect to valsartan. In this case, the changes in the SBP are probably the most rational explanation for the interpretation of the results. However, LCZ696 recovered the normal histological structure of the myocardiocytes, so we can assume that the growth factors involved in this process could also undergo downregulation, probably starting from a reduction in inflammation as we will see later [22,23].
On the other hand, the FFHR showed vascular remodelling. This change is probably the most difficult to reverse in the experimental model because it depends more on the underlying inflammatory process than on the hemodynamic changes, as have seen in other previously published works. LCZ696 modified the vascular structure, showing clearly significant differences with respect to FFHR, like Valsartan. However, vascular inflammation was reduced. When study the IL-6Ralfa pathway, was see that FFHR showed a great increase in the expression of STAT3, with a significant increase in receptor expression. LCZ696 significantly decreased the expression of STAT3, even more than valsartan. That demonstrated a direct effect on the activation pathway of hsCPR, which also decreased significantly with respect to FFHR. Although the expression of the STAT3 phosphorylated could not be demonstrated, its direct post-transcriptional product, hsPCR, was normalized with LCZ696 [24-26].
This could also be explained because the main STAT3 phosphorylator is JAK, which can be activated through AT1R and IL-6R or directly by oxidative stress, presumably this protein is also reduced in the treatment with LCZ696, because was showed significant differences with respect to V [27-29].
We think that the LCZ696 could modify the damage of the target organ, even more significantly than the single block of AT1R. So, could LCZ could modify vascular repair? To answer this question, we evaluated the presence of EPCs, in an advanced stage of maturation (VEGFR2 + / CD133 +) at the vascular endothelium level. The FFHR model does not have mature EPC at the endothelial level, as we could see in this study from the expression of these markers by WB and IHC. When the model received, LCZ696 completely reversed and showed a similar pattern of expression as the rats in the control group. This effect was exclusive to LCZ696. This is the most important finding of our work because this action has not been previously described [30,31].
Finally, it was decided to evaluate the expression of MAS1R at vascular level, since this receptor could increase the expression of VEFGR2 that occurs at the endothelial level favouring endothelial repair. In the present study, we analysed the expression of VEFGR2 with CD133, these molecules originate de novo from the formation of endothelial cells. The expression of MASR1 increased significantly, was treated with LCZ696 but not with valsartan. This could corroborate the hypothesis that, the action of LCZ696 on MASR1, produce an increase of EPC at the endothelial level [32,33].
The Phyto pathological mechanisms proposed in our work are summarized in Figure 3. The dual blocking of NEP/AT1R by LCZ696 could reduce the expression and phosphorylation of STAT3 through JAK, either by blocking AT1R, reducing oxidative stress or controlling systolic blood pressure.
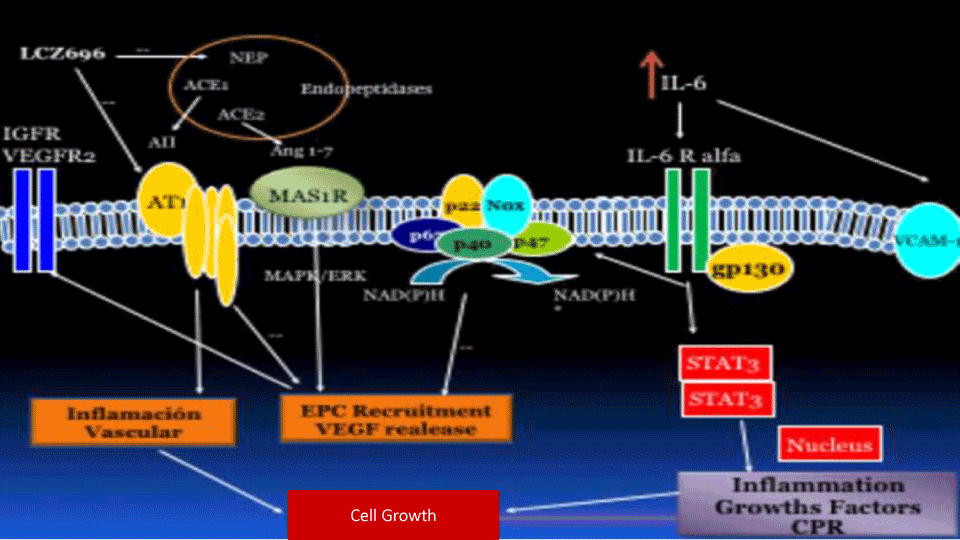
Figure 3. The phytopathological mechanisms proposed in our work are summarized in this image. The dual blocking of NEP/AT1R by LCZ696 could reduce the expression and phosphorylation of STAT3 through JAK, either by blocking AT1R, reducing oxidative stress or controlling systolic blood pressure. The reduction of STAT3 produced a decrease in the inflammatory transcription factors in the nucleus and a release of hsCPR in the blood circulation, which produces an increasing docking of the alpha subunit of IL6-R towards the membrane. On the other hand, NEP and ACE2 probably induce the conversion of angiotensin II in different intermediate metabolites, such as angiotensin 1-7 (Ang 1-7), which produces antagonistic effects to angiotensin II by MAS1R. The intracellular cascade of MAS1R, by MAPK / ERK, produces as a fundamental effect, namely the production of VEGF and its two receptors: VEGFR1 and 2. From this mechanism, the endothelium could be repaired and/or replaced, favouring the maturation of circulating EPCs on resident EPCs at the endothelial level. MAS1R could be counter-regulated by IL-1ß
The reduction of STAT3 produced a decrease on inflammatory transcription factors in the nucleus and a release of hsCPR in the blood circulation, which produces an increasing docking of the alpha subunit of IL6-R towards the membrane. Through this pathway, vascular remodelling and LVH were reduced because part of the growth factors and migration of muscle cells depends on the activation of the inflammatory cascade.
On the other hand, NEP and ACE2 probably induce the conversion of angiotensin II in different intermediate metabolites, such as angiotensin 1-7 (Ang 1-7), which produces antagonistic effects to angiotensin II by MAS1R. The intracellular cascade of MAS1R, by MAPK / ERK, produces as a fundamental effect, namely the production of VEGF and its two receptors: VEGFR1 and 2. From this mechanism, the endothelium could be repaired and/or replaced, favouring the maturation of circulating EPCs on resident EPCs at the endothelial level. MAS1R could be counter-regulated by IL-1ß [34-36].
In conclusion, we postulate that LCZ696, by MAS1R activation, is not only able to improve endothelial function but is able to repair the endothelium, and this probably allows for improved functionality of the entire cardiovascular system. In addition, LCZ696 could reduce the expression of hsCPR through reduction in the expression of STAT3, a sign also demonstrated in different clinical studies such as JUPITER and CANTOS, which have allowed a great reduction in morbidity and mortality, revolutionizing modern cardiology. The anti-inflammatory and angio-repairing effect of LCZ696 is probably reflected in an improvement in the survival of patients who receive a treatment regimen with this drug in studies such as PARADIGH-HF [37-39].
The authors do not have any conflicts of interest in the manuscript.
- This work was carried out with the funding granted by the National University of Cuyo in its call SIIP Projects 2016-2018.
- Subsidies for scientific divulgation for students of PhD in medicine UNCuyo 2018.
- Eugene Braunwald (2015) the path to an angiotensin receptor antagonist-neprilysin inhibitor in the treatment of heart failure. J Am Coll Cardiol 65: 1029-1041.
- Chirag B, Franz HM, Bernard K, Luis MR, Kazuomi K (2015) Role of neprilysin inhibitor combinations in hypertension: insights from hypertension and heart failure trials. Eur Heart J 36:1967-1973.
- Thomas GL, Bing HW, Andrew RK, Li H, Randy W, et al. (2015) angiotensin receptor neprilysin inhibitor lcz696 attenuates cardiac remodeling and dysfunction after myocardial infarction by reducing cardiac fibrosis and hypertrophy. Circ Heart Fail 8: 71-78.
- Desai AS, McMurray JJ, Packer M (2015) Effect of the angiotensin-receptor-neprilysin inhibitor LCZ696 compared with enalapril on mode of death in heart failure patients. Eur Heart J 36: 1990-1997.
- Renna NF, Diez ER, Lembo C, Miatello RM (2013) Role of Cox-2 in vascular inflammation: an experimental model of metabolic syndrome. Mediators Inflamm 2013: 513251.
- Renna NF, Diez EA, Miatello RM (2014) Effects of dipeptidyl-peptidase 4 inhibitor about vascular inflammation in a metabolic syndrome model. PLoS One 9: e106563.
- Renna NF, de Las Heras N, Miatello RM (2013) Pathophysiology of vascular remodeling in hypertension. Int J Hypertens 2013: 808353. [Crossref]
- Renna NF, Lembo C, Diez E, Miatello RM (2013) Role of Renin-Angiotensin system and oxidative stress on vascular inflammation in insulin resistence model. Int J Hypertens 2013: 420979.
- Renna NF (2013) Oxidative stress, vascular remodeling, and vascular inflammation in hypertension. Int J Hypertens 2013: 710136. [Crossref]
- Kishimoto T (2010) IL-6: from its discovery to clinical applications. Int Immunol 22: 347-352. [Crossref]
- Rose-John S (2001) Coordination of interleukin-6 biology by membrane bound and soluble receptors. Adv Exp Med Biol 495: 145-151. [Crossref]
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, et al. (2003) Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J 374: 1-20. [Crossref]
- Rosenzwieg A (2005) Circulation endothelial progenitors-cells as biomarkers. N Engl J Med 353: 1055-1056.
- Basile DP, Mervin C (2014) Circulating and tissue resident endothelial progenitor cells. J Cell Physiol 229: 10-16.
- Fadini GP, Miorin M, Facco M, Bonamico S, Baesso I, et al. (2005) Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol 45: 1449-1457.
- Fadini GP, Coracina A, Baesso I, Agostini C, Tiengo A, et al. (2006) Peripheral blood CD34+KDR+ endothelial progenitor cells are determinants of subclinical atherosclerosis in a middle-aged general population. Stroke 37: 2277-2282.
- Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, et al. (2005) Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med 353: 999-1007. [Crossref]
- Zanchetti A, Thomopoulos C, Parati G (2015) Randomized controlled trials of blood pressure lowering in hypertension: a critical reappraisal. Circ Res 13: 116:1058-1073.
- Ushijima K, Takuma M, Ando H, Ishikawa-Kobayashi E, Nozawa M, et al. (2013) Effects of telmisartan and valsartan on insulin sensitivity in obese diabetic mice. Eur J Pharmacol 698: 505-510. [Crossref]
- von Lueder TG, Wang BH, Kompa AR, Huang L, Webb R, et al. (2015) Angiotensin receptor neprilysin inhibitor LCZ696 attenuates cardiac remodeling and dysfunction after myocardial infarction by reducing cardiac fibrosis and hypertrophy. Circ Heart Fail 8:71-78.
- Gradman AH (2015) LCZ696: the next step in improving RAS inhibition? Curr Hypertens Rep 17: 37. [Crossref]
- Schmieder RE, Wagner F, Mayr M, Delles C, Ott C, Keicher C, et al. (2017) The effect of sacubitril/valsartan compared to olmesartan on cardiovascular remodeling in subjects with essential hypertension: the results of a randomized, double-blind, active-controlled study. J Eur Heart J 38: 3308-3317.
- Wong PC, Guo J, Zhang A2 (2017) The renal and cardiovascular effects of natriuretic peptides. Adv Physiol Educ 41: 179-185. [Crossref]
- Tanaka T, Narazaki M, Kishimoto T (2016) Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy 8: 959-970.
- Ridker PM (2016) From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream To Identify Novel Targets for Atheroprotection. Circ Res 118: 145-156.
- Anderson DR, Poterucha JT, Mikuls TR, Duryee MJ, Garvin RP, et al. (2013) IL-6 and its receptors in coronary artery disease and acute myocardial infarction. Cytokine 62: 395-400. [Crossref]
- Vogel TP, Milner JD, Cooper MA4 (2015) The Ying and Yang of STAT3 in Human Disease. J Clin Immunol 35: 615-623. [Crossref]
- Haghikia A, Ricke-Hoch M, Stapel B, Gorst I, Hilfiker-Kleiner D (2014) STAT3, a key regulator of cell-to-cell communication in the heart. Cardiovasc Res 102: 281-289. [Crossref]
- Lim CP, Fu XY (2012) Multiple roles of STAT3 in cardiovascular inflammatory responses. Prog Mol Biol Transl Sci 106: 63-73. [Crossref]
- Zhang M, Malik AB, Rehman J (2014) Endothelial progenitor cells and vascular repair. Curr Opin Hematol 21: 224-228. [Crossref]
- Kienstra KA, Hirschi KK (2012) Vascular progenitor cell mobilization. Methods Mol Biol 904: 155-164. [Crossref]
- Cerniello FM1, Carretero OA1, Longo Carbajosa NA1, Cerrato BD1, Santos RA1, et al. (2017) MAS1 Receptor Trafficking Involves ERK1/2 Activation Through a β-Arrestin2-Dependent Pathway. Hypertension 70: 982-989. [Crossref]
- Khajah MA, Fateel MM, Ananthalakshmi KV, Luqmani YA (2016) anti-inflammatory action of angiotensin 1-7 in experimental colitis. PLoS One 11: e0150861. [Crossref]
- Brian RH, Timothy JS, Jordan RW, Daniela ND, Eric CE, et al. (2017) Mechanisms of mas1 receptor-mediated signaling in the vascular endothelium. Arterioscler Thromb Vasc Biol 37: 433-445.
- Fraga-Silva RA, Da Silva DG, Montecucco F, Mach F, Stergiopulos N, et al. (2012) The angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas receptor axis: a potential target for treating thrombotic diseases. Thromb Haemost 108: 1089-1096.
- Ridker PM (2013) Closing the loop on inflammation and atherothrombosis: why perform the CIRT and CANTOS trials? Trans Am Clin Climatol Assoc 124: 174-190. [Crossref]
- Vidt DG, Ridker PM, Monyak JT, Schreiber MJ, Cressman MD (2011) Longitudinal assessment of estimated glomerular filtration rate in apparently healthy adults: a post hoc analysis from the JUPITER study (justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin). Clin Ther 33: 717-725. [Crossref]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, et al (2017) Anti-inflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377: 1119-1131.
- Simpson J, Jhund PS, Silva Cardoso J, Martinez F, Mosterd A, et al. (2015) Comparing LCZ696 with enalapril according to baseline risk using the MAGGIC and EMPHASIS-HF risk scores: an analysis of mortality and morbidity in PARADIGM-HF. J Am Coll Cardiol 66: 2059-2071.