“Novelty and Impact”: Genome wide methylation analysis of cervical swabs of a cohort of cervical intraepithelial neoplasia (CIN I-III) and cervical cancer patients revealed a large number of genes with hypermethylated promoter, and a markedly lower number hypomethylated, in CIN III and cervical cancer. Our results support the prognostic value of methylation profiling of a larger number of genes than previously thought. These can be readily identified in swabs rather than biopsies, which facilitates their clinical use as prognostic markers.
Cancer of the uterine cervix is caused by a subset of oncogenic human Papillomavirus (HPV)-types with mucosal tropism. Besides the known effects of the viral oncoproteins on cellular functions there is evidence suggesting that cervical carcinogenesis involves epigenetic changes in the host DNA. In this study, we have examined the global promoter methylation profile associated with progression to cervical cancer at wide genome scale. The methylation pattern of nearly 14,000 genes was analyzed in cervical swabs at different stages of cervical carcinogenesis: low-grade cervical intraepithelial neoplasia (CIN I and II), high-grade CIN (CIN III) and invasive cancer, as well as healthy individuals. Unsupervised analysis (Hierarchical Clustering) identified two groups: A) healthy, CIN I and CIN II; and B) CIN III/cancer. Supervised T-Test analysis showed 1069 promoter regions hypermethylated and 85 hypomethylated in CIN III/cancer compared to CIN I/CIN II and healthy samples (p<0.0001). Overall, the differentially methylated genes act in transcription, cell cycle, apoptosis and cell adhesion pathways. Of the hypermethylated genes, 132 (12.3%) were down-regulated in a matched cervical cancer group. In turn, only 4 (4.7%) of the hypomethylated genes were overexpressed in that group. These data suggest that, although significant changes in methylation of a large number of cellular promoters take place during progression to cervical cancer, correlation between methylation and altered gene expression occurs only in a reduced number of genes. Nevertheless, these genes are relevant to cell transformation. Our results support the prognostic value of methylation profiling of a larger number of genes than previously thought.
cervical cancer, cervical intraepithelial neoplasia (cin), epigenetics, genome-wide dna-methylation, gene expression, human papillomavirus (hpv)
Cervical cancer (CC) of the uterus is the third most frequent cancer in women worldwide with over half a million new cases estimated in the year 2012 [1]. Over 85% of the cases occur in developing countries. In Colombia, the estimated incidence of CC is 21.5 cases per 100.000 inhabitants [1]. The cervical carcinogenesis begins with non-invasive dysplastic lesions, which progress to cervical intraepithelial neoplasia (CIN) grades I to III and, in some cases, the lesions persist and progress to invasive carcinoma [2]. A few high-risk human papillomaviruses (HPVs), most notably HPV16 and HPV18, are in the etiology of the majority of CCs, as well as other anogenital tumors and an increasing number of head and neck cancers [3]. Two proteins encoded by these viruses, E6 and E7 have transforming capabilities by inactivating RB1 and promoting degradation of TP53 thereby interfering with signaling pathways and cell cycle control mechanisms [4]. However, the natural history of the disease shows that only a minority of women infected with high-risk HPVs cannot clear infection and develop CC [5]. The molecular mechanisms that determine clearance or persistence of the HPV infection are mainly unknown. Thus, to date there are no reliable molecular markers that may help predicting whether low-grade lesions will progress to cancer or rather regress and be cleared.
Cancer cells develop their malignant phenotype through a series of concurrent genetic and epigenetic changes that cooperatively influence gene transcription and contribute to overall malignant transformation [6]. The epigenetic machinery plays an important role in the control of the transcriptional activity through DNA modifications, most importantly CpG cytosine-5 methylation [6]. The human genome can be envisioned as immense regions of DNA sequence containing methylated CpG dinucleotides distributed randomly (CpG oceans), short regions of higher density, unmethylated CpG dinucleotides (CpG islands), and transition regions (CpG shores) with conserved tissue-specific methylation [7]. CpG islands are present in the promoter regions of nearly half of the actively transcribed human genes. They are unmethylated in normal cells, but often hypermethylated in cancer cells, and this is an epigenetic change typically associated with transcriptional repression. From a functional point of view, the presence of methyl groups on cytosines can result in the local alteration of the structure of chromatin by the union of binding proteins to methyl-cytosine, chromatin remodeling proteins and histone deacetylases, all of which hinder recognition of promoter sequences by the cellular transcription machinery. Besides, it is conceivable that methylation patterns characteristic of cancer cells could be identified in early stages of carcinogenesis, and hence methylation biomarkers may help in early detection and/or prognostic assessment of CC.
The use of high-performance platforms for evaluating profiles of methylation allows detection of large numbers of methylated genes that are possibly associated with the tumorigenesis process. Currently there are several methodologies to determine the methylation profile of genomic DNA samples. In a general way, these methods include [8]: (i) DNA digestion with methylation-sensitive restriction enzymes, combined bisulfite restriction analysis (COBRA), methylation-specific PCR (MSP), which require at least 1-10 ug per sample; (ii) methodologies based on proteins or antibodies that can bind to the methyl group, which have the disadvantage of allowing the processing of only limited numbers of samples in parallel; and (iii) genome-wide DNA methylation profiling, such as the Illumina Infinium platform, that generally require from 500ng to 1ug of DNA and allows the processing of larger numbers of samples simultaneously.
In cervical cancer cells carrying one or more oncogenic HPV types de novo CpG methylation occurs in both viral and host DNA. It has been shown that the DNA of HPV16 and HPV18 in pre-immortal keratinocytes is almost completely unmethylated while it is heavily methylated in their immortalized derivative cells [9]. In addition, these studies showed increasing hypermethylation of the CpG islands in the HPV DNA during progression from dysplasia to invasive cancer. On the other hand, analyses of the methylation status of the host genome have shown that the promoter region of over 50 genes is hypermethylated in cervical cancers as well as their exfoliated cells [10]. At least 15 of these genes were studied also in CIN2 and CIN3 and showed intermediate methylation levels. Only a few of these genes were analyzed in several independent studies [10], most of which used MSP, quantitative MSP (qMSP), bisulfite sequencing or methylation-sensitive restriction analysis. The most significant genes found hypermethylated in cervical cancer in previous studies were CDH1, MAL, TFP12, DAPK1, RARB and TWIST1[11]. A panel including the last three genes was claimed to show high sensitivity and specificity for early detection of cervical cancer. However, a meta-analysis of 51 studies on methylation in cervical cancer showed that there is a high variability in the rate of methylation of a given gene among different studies [11].
Therefore, there is still a need for extensive, genome-wide methylation studies that may help elucidate the role of epigenetic modifications in progression from dysplastic lesions to cervical cancer and find methylation biomarkers with prognostic value. The use of microarrays allows simultaneous assessment of the methylation status of virtually any gene in cervical cancer and CIN in comparison with normal tissue. In the present study we have used the Illumina Human-Methylation27 Infinium BeadChip to compare the genome-wide DNA methylation profiles of low and high-grade CIN and cervical cancer as well as normal cervix exfoliates. Supervised T-Test analysis of the methylation patterns in CIN and cancer exfoliates identified an overwhelming number of hypermethylated gene promoters in contrast to a reduced number of hypomethylated genes. Further analyses of data on expression of those genes revealed discrepancy in the correlation between methylation status and expression levels.
Patients and samples
This study is based on a descriptive, cross-sectional and observational analysis. All tissue samples were obtained after signed informed consent of all participating patients and healthy donors with approval of the institutional review board. The number of patients included in this study was calculated according to biometric parameters. Cervical specimens were collected over a period of 3 months in a hospital (Dinamica-IPS) of the city of Bogotá. Trained medical personnel took the cervical swabs. The cohort of patients included 3 women with no signs of cervical pathology, and 17 patients with confirmed cervical histopathologic diagnosis, namely: 4 with CIN I, 4 with CIN II, 3 with CIN III, 3 with in situ squamous cell carcinoma (SCC) and 3 with invasive SCC. None of these patients had been subjected to treatment before the cervical swabs were collected.
The cervical swabs for this study were deposited in PBS containing 0.05% thimerosal. DNA was isolated from these samples by phenol-chloroform extraction and subsequently used for bisulfite conversion and methylation analyses by BeadChip microarray (see below) and for multiplex HPV genotyping (see below). The quality control of the samples was performed by spectrophotometric measuring at OD260/230 and OD260/280 within a range of 1.0-3.0 and a concentration of approximate 1500ng/μL using a Nanodrop1000®.
Bisulfite conversion of genomic DNA
The bisulfite conversion of genomic DNA was performed using the EZ Methylation Kit (Zymo Research, D5002), following the manufacturer's instructions. Briefly, DNA concentration was measured by fluorescence using Quant-iT™ PicoGreen ®, and 1μg of DNA was mixed with a buffer solution and incubated at 37°C for 15 minutes, subsequently mixed with the conversion reagent and incubated in a thermocycler for 16 cycles at 95°C for 30 seconds and 50°C for 60 minutes. Finally, the samples were converted, desulphonated and purified according to the manufacturer's recommendations.
Human Infinium Methylation27 Illumina BeadChip
The converted genomic DNA was analyzed by Illumina Infinium BeadChip Methylation27 Human Kit at the Made-Radiumhospitalet Rikshospitalet Medical Center / University of Oslo Microarray Core Facility (Norwegian Microarray Consortium). The BeadChip assay provides quantitative measurements of DNA methylation for 27578 CpG dinucleotides covering the promoter region of 14,495 human genes. Chip data processing and analysis was performed using Illumina reagents and following the manufacturer's instructions. The procedure began with DNA denaturation converted with 0.014N sodium hydroxide, neutralized and amplified for 20-24 hours at 37°C. The samples were hybridized to the BeadChip at 48°C for 16-20 hours. After incubation, the beadchips were washed with buffer and placed in the Fluidics Station finally the image was captured on the Illumina iScan scanner. The data were obtained using the BeadStudio v3.0 software. The heatmap was obtained with the Multiexperiment Viewer software.
Multiplex HPV Genotyping (MPG)
Multiplex BSGP5+/GP6+ PCR-MPG was carried out with primers reported previously [12] essentially as described [13]. The assay used nine sense primers BSGP5+ at 200 nM and 3 antisense primers 5'-biotinylated-BSGP6+ at 400 nM to amplify a ~150 bp fragment of the viral L1 ORF, as well as two additional primers (MS3/MS10) at 300 nM to amplify the ß-globin gene, which was used as DNA integrity control. The multiplex Luminex hybridization assay enabled the detection of 27 mucosal HPV-genotypes: i) high risk (HR)-HPV types: 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68; ii) possibly HR-HPV types: 26, 53, 67, 70, 73, 82; iii) low risk (LR)-HPV types: 6, 11, 30, 43, 44 and 69. In addition, the β-globin product was amplified in each sample.
Oligonucleotide probes with an amino group at the 5' end for each of the 27 HPV types were coupled to carboxylated beads using the carbodiimide procedure as described previously [14]. For each combination of probes and beads we used 2.5 million carboxylated beads in 25 µL of 0.1 M2-(N-morpholino) ethanesulfonic acid (MES), pH 4.5, with 200 µg of N-(3-dimethylaminopropyl)-N-ethylcarbodiimide (EDC) and 400 pmol of probe, the mixture was incubated in the dark under stirring for 30 minutes, the addition of EDC and incubation was repeated once, then the beads were washed with 0.5 ml of 0.2 g/L of Tween 20 and once with 0.5 ml of 1.0 g/L SDS. Finally, they were stored at 4°C in TE solution. Hybridization was performed in 96 well plates with 10 µL of PCR product in 33 µL of hybridization solution (0.15 M of tetramethylammonium chloride (TMAC), 75 mM Tris-HCl, 6 mM EDTA, 1.5 g/L Sarkosyl, pH 8.0) to which 2000 bead-coupled probes were added. This mixture was incubated at 95°C for 10 minutes and immediately placed on ice for 2 minutes, then it was placed at 41°C for 30 minutes under stirring, then the samples were transferred to a wash plate with filter bottom (Millipore, Bedford, MA) pre-equilibrated with blocking solution (PBS with 0.2 g/L Tween-20 and 2.0 M TMAC), and washed with blocking solution to eliminate non-hybridized DNA in a vacuum wash station (Millipore). Subsequently, biotinylated PCR products were stained with streptavidin-R-phycoerythrin conjugate (Molecular Probes) diluted 1:1600 in 2.0 M TMAC, 75 mM Tris-HCl, 6 mM EDTA, 1.5 g/L Sarkosyl, 1.0 g/L casein, pH 8.0 and incubated 20 minutes at room temperature with shaking. After washing three times with blocking solution, the beads were analyzed in a Luminex 100 reader (Luminex Corp.), which uses two lasers, one that recognizes the bead set by the internal bead color and another to quantify the reporter fluorescence on the bead. The results were expressed in median fluorescence intensities (MFI) of ≥100 beads per sample [12,14].
HPV16 and HPV18 transcript copy number
We analyzed the expression of the early gene E7 of HPV16 and HPV18. To this end, we isolated total RNA from biopsy material included in paraffin with the kit RNeasy FFPE (Qiagen®). After DNase I treatment and reverse transcription with the kit “High Capacity RNA-to-cDNA” (Applied Biosystem®) and analyzed them by quantitative real time PCR (qRT-PCR) using specific primers for E7 and E6/E7 transcripts (Supplementary Table 1). The estimation of the transcript copy number was made by absolute quantification using known amounts of HPV-16 and HPV-18 DNA to generate a standard curve.
Table 1. Data on selected genes with higher degree of methylation, irrespective of their expression status, in cervical swabs of CIN III/cervical cancer patients compared with CIN I/CIN II patients and healthy donors.
CpG ID |
Gen |
Description |
RefSeq |
Entrez |
Group A |
Group B |
∆-ß |
Fold
Change |
Adjusted
p value |
Mean |
SD |
Mean |
SD |
cg23349790 |
IGSF21 |
immunoglobin superfamily member 21 |
NM_032880.2 |
84966 |
0.10 |
0.08 |
0.70 |
0.19 |
0.59 |
6.72 |
5.60E-6 |
cg09053680 |
UTF1 |
undifferentiated embryonic cell transcription factor 1 |
NM_003577.2 |
8433 |
0.06 |
0.08 |
0.63 |
0.20 |
0.57 |
10.44 |
1.21E-5 |
cg09516965 |
PTGDR |
prostaglandin D2 receptor |
NM_000953.2 |
5729 |
0.17 |
0.28 |
0.72 |
0.21 |
0.55 |
4.13 |
2.07E-4 |
cg08185661 |
SYT9 |
synaptotagmin 9 |
NM_175733.2 |
143425 |
0.09 |
0.08 |
0.63 |
0.17 |
0.54 |
7.38 |
5.81E-6 |
cg04534765 |
GALR1 |
galanin receptor 1 |
NM_001480.2 |
2587 |
0.10 |
0.08 |
0.64 |
0.19 |
0.54 |
6.49 |
1.38E-5 |
cg01009664 |
TRH |
thyrotropin releasing hormone |
NM_007117.1 |
7200 |
0.12 |
0.08 |
0.65 |
0.13 |
0.54 |
5.69 |
1.83E-7 |
cg17861230 |
PDE4C |
phosphodiesterase 4C |
NM_000923.1 |
5143 |
0.15 |
0.08 |
0.69 |
0.14 |
0.54 |
4.58 |
2.45E-7 |
cg21458907 |
CADPS |
calcium dependent secretion activator |
NM_183393.1 |
8618 |
0.06 |
0.04 |
0.59 |
0.18 |
0.54 |
10.29 |
2.69E-5 |
cg25437385 |
SLC35F3 |
solute carrier family 35 member F3 |
NM_173508.1 |
148641 |
0.08 |
0.05 |
0.62 |
0.11 |
0.54 |
7.58 |
2.83E-8 |
cg00489401 |
FLT4 |
fms related tyrosine kinase 4 |
NM_182925.1 |
2324 |
0.10 |
0.06 |
0.64 |
0.19 |
0.53 |
6.13 |
1.83E-5 |
cg18443378 |
WDR17 |
WD repeat domain 17 |
NM_181265.2 |
116966 |
0.08 |
0.09 |
0.61 |
0.17 |
0.53 |
7.38 |
5.36E-6 |
cg11657808 |
RYR2 |
ryanodine receptor 2 |
NM_001035.1 |
6262 |
0.11 |
0.10 |
0.64 |
0.20 |
0.53 |
5.69 |
2.36E-5 |
cg08453021 |
ELMO1 |
engulfment and cell motility 1 |
NM_014800.8 |
9844 |
0.04 |
0.02 |
0.56 |
0.15 |
0.51 |
12.65 |
6.10E-6 |
cg08575537 |
EPO |
erythropoietin |
NM_000799.2 |
2056 |
0.16 |
0.10 |
0.67 |
0.16 |
0.51 |
4.29 |
1.67E-6 |
cg07017374 |
FLT3 |
fms related tyrosine kinase 3 |
NM_004119.1 |
2322 |
0.09 |
0.09 |
0.59 |
0.19 |
0.51 |
6.93 |
1.79E-5 |
cg11438428 |
PTF1A |
pancreas specific transcription factor, 1a |
NM_178161.1 |
256297 |
0.12 |
0.05 |
0.62 |
0.15 |
0.51 |
5.26 |
4.75E-6 |
cg26609631 |
GSX1 |
GS homeobox 1 |
NM_145657.1 |
219409 |
0.12 |
0.10 |
0.62 |
0.15 |
0.50 |
5.30 |
1.10E-6 |
cg02164046 |
SST |
somatostatin |
NM_001048.3 |
6750 |
0.08 |
0.03 |
0.58 |
0.20 |
0.50 |
6.91 |
6.56E-5 |
cg26128092 |
WRAP73 |
WD repeat containing, antisense to TP73 |
NM_017818.2 |
49856 |
0.09 |
0.05 |
0.58 |
0.17 |
0.50 |
6.77 |
1.32E-5 |
cg00027083 |
EPB41L3 |
erythrocyte membrane protein band 4.1 like 3 |
NM_012307.2 |
23136 |
0.13 |
0.14 |
0.62 |
0.12 |
0.49 |
4.73 |
3.89E-7 |
cg13877915 |
ZNF132 |
zinc finger protein 132 |
NM_003433.2 |
7691 |
0.05 |
0.07 |
0.54 |
0.20 |
0.49 |
10.02 |
1.52E-4 |
cg22619018 |
CSMD1 |
CUB and Sushi multiple domains 1 |
NM_033225.3 |
64478 |
0.11 |
0.09 |
0.60 |
0.24 |
0.49 |
5.53 |
2.99E-4 |
cg22881914 |
NID2 |
nidogen 2 |
NM_007361.2 |
22795 |
0.13 |
0.14 |
0.61 |
0.24 |
0.49 |
4.81 |
2.10E-4 |
cg19831575 |
FGF4 |
fibroblast growth factor 4 |
NM_002007.1 |
2249 |
0.08 |
0.06 |
0.57 |
0.24 |
0.48 |
6.90 |
3.71E-4 |
cg01805540 |
CACNB2 |
calcium voltage-gated channel auxiliary subunit beta 2 |
NM_201593.1 |
783 |
0.07 |
0.06 |
0.55 |
0.20 |
0.48 |
8.44 |
6.42E-5 |
cg17371081 |
NELL1 |
neural EGFL like 1 |
NM_006157.2 |
4745 |
0.05 |
0.04 |
0.54 |
0.25 |
0.48 |
10.39 |
4.37E-4 |
cg03544320 |
CRMP1 |
collapsin response mediator protein 1 |
NM_001313.3 |
1400 |
0.25 |
0.19 |
0.73 |
0.10 |
0.48 |
2.95 |
7.08E-6 |
cg25302419 |
CTNND2 |
catenin delta 2 |
NM_001332.2 |
1501 |
0.07 |
0.07 |
0.56 |
0.18 |
0.48 |
7.55 |
2.37E-5 |
cg07533148 |
TRIM58 |
tripartite motif containing 58 |
NM_015431.2 |
25893 |
0.06 |
0.06 |
0.54 |
0.25 |
0.48 |
8.76 |
5.59E-4 |
cg09551147 |
SORCS3 |
sortilin related VPS10 domain containing receptor 3 |
NM_014978.1 |
22986 |
0.05 |
0.04 |
0.53 |
0.24 |
0.47 |
9.89 |
3.78E-4 |
cg12128017 |
EPHA10 |
EPH receptor A10 |
NM_173641.1 |
284656 |
0.08 |
0.05 |
0.55 |
0.18 |
0.47 |
7.15 |
6.94E-5 |
cg20291049 |
POU3F3 |
POU class 3 homeobox 3 |
NM_006236.1 |
5455 |
0.16 |
0.09 |
0.64 |
0.18 |
0.47 |
3.88 |
1.74E-5 |
cg19096475 |
CLMP |
CXADR like membrane protein |
NM_024769.2 |
79827 |
0.11 |
0.05 |
0.58 |
0.14 |
0.47 |
5.27 |
2.89E-6 |
cg11389172 |
SLC18A3 |
solute carrier family 18 member A3 |
NM_003055.1 |
6572 |
0.05 |
0.04 |
0.52 |
0.24 |
0.47 |
11.29 |
3.67E-4 |
cg10303487 |
DPYS |
dihydropyrimidinase |
NM_001385.1 |
1807 |
0.09 |
0.07 |
0.56 |
0.20 |
0.47 |
6.31 |
8.38E-5 |
cg19064258 |
HS3ST2 |
heparan sulfate-glucosamine 3-sulfotransferase 2 |
NM_006043.1 |
9956 |
0.11 |
0.06 |
0.58 |
0.18 |
0.47 |
5.14 |
3.08E-5 |
cg27403635 |
KCNN2 |
potassium calcium-activated channel subfamily N member 2 |
NM_021614.2 |
3781 |
0.09 |
0.09 |
0.56 |
0.15 |
0.47 |
6.14 |
3.51E-6 |
cg15747595 |
TSPYL5 |
TSPY like 5 |
NM_033512.2 |
85453 |
0.14 |
0.08 |
0.60 |
0.15 |
0.47 |
4.44 |
2.12E-6 |
cg06722633 |
GRIK3 |
glutamate ionotropic receptor kainate type subunit 3 |
NM_000831.2 |
2899 |
0.12 |
0.10 |
0.59 |
0.21 |
0.47 |
4.80 |
7.96E-5 |
cg17525406 |
AJAP1 |
adherens junctions associated protein 1 |
NM_018836.2 |
55966 |
0.12 |
0.08 |
0.59 |
0.24 |
0.47 |
4.89 |
4.04E-4 |
Statistical and bioinformatic analyses
Statistical analyses and heat map visualization were performed with the MultiExperiment Viewer software (MeV 4.6) [15], with an unsupervised cluster analysis with Euclidian distance to create a heatmap. This analysis showed that there was a turning point in gene methylation between the group of negative, CIN I and CIN II samples, and the group of CIN III, in situ carcinoma and invasive cancer. The Benjamini and Hochberg method [16] was used to adjust the p-values for multiple testing. The selection of differentially methylated genes was performed with T-test p = 0.0001. For automated functional annotation and gene enrichment analysis, we used the Database for Annotation, Visualization and Integrated Discovery (http://david.niaid.nih.gov/david) [17]. The DAVID resource calculates over-representation of specific biological themes/pathways with respect to the total number of genes assayed and annotated. The REVIGO resource was employed to summarize and visualize the enriched GO terms in an interactive graph based on the p-values obtained by DAVID [18].
The microarray analysis of gene expression used in this study was the Affymetrix HG-U133A platform and Gene Expression Omnibus (GEO) (URL: www.ncbi.nlm.nih.gov/geo) accession number GSE7803 [19]. The selection of differentially expressed genes was performed with an unsupervised cluster analysis with Euclidian distance and p = 0.0001. Finally, a correlation of hypermethylation and down-regulation of expression was performed using contingency tables.
The dataset generated in this study is available and can be retrieved from the Gene Expression Omnibus (GEO; www.ncbi.nlm.nih.gov/geo/) accession number GSE41384.
Selection of patients, HPV detection and genotyping
Previous studies have identified cancer-specific DNA methylated regions and it has been suggested that increased methylation variability involves loss of epigenetic stability of well-defined genomic domains [20]. In addition, algorithms are being developed that could help predict progression of precursor lesions to invasive cancer [21]. Studies on differential DNA methylation are expected to identify sets of genes that, according to their methylation status, could determine the altered expression of their encoded proteins in cervical cancer and its precursor lesions compared with normal cervical tissue. In addition, their changes in methylation during cancer progression could be of prognostic value. The present study was designed to analyze methylation profiles in DNA purified from cervical exfoliates taken from CIN and cervical cancer patients, and healthy donors before the histopathologic diagnosis was made. Such samples offer the advantage of being easily accessible and do not require invasive techniques for their collection. In contrast, methods based on the analysis of DNA obtained directly from cervical dysplastic tissue require taking biopsy samples, which can have local side effects such as hemorrhage and, eventually, could contribute to spread the initial lesion to underlying tissues.
The DNA samples investigated in this study were purified from cervical swabs of 20 donors who were subsequently diagnosed of CIN I (n=3), CIN II (n=4), CIN III, carcinoma in situ (n=6) and invasive cervical cancer (n=3). Our study included also 3 cases with no detectable cervical pathology (healthy donors, n=3), although they had a positive HPV test. The socio-demographic and clinical data of the study population relevant to the main risk factors associated with cervical cancer carcinogenesis, such as age, number of sexual partners, number of pregnancies and history of genital infection are summarized in the supplementary table 2. The presence of HR and LR-HPV types in the cervical swabs was analyzed by multiplex PCR genotyping (MPG), a highly sensitive technique described elsewhere [13]. The results are shown in the supplementary table 3. HPV was detected in all the patients as well as the healthy donors participating in the study. HPV16 and HPV18 were the most prevalent virus types (n=19, 95%). In 3 cases these viruses co-existed with LR-HPV types and only one case was associated with HR-HPV types other than HPV16 and HPV18. The number of copies of the respective E7 transcripts was estimated by quantitative RT-PCR. In the samples of all healthy donors and in nearly half of the patients the number of copies was below 50. In the rest the copy number ranged from 50-100 (Supplementary table 3).
Table 2. Data on selected genes whose promoter regions had a higher degree of methylation and their expression was diminished in CIN III/cervical cancer compared with CIN I/CIN II and healthy donor samples. The table shows the 40 genes with the highest degree of methylation (Ƨ-values between 0.59 and 0.47, fold change between 9.74 and 4.06) and the lowest expression level (fold change between -2.18 and -2.07) in CIN III and cervical cancer compared to normal cervical tissue.
Gene |
Description |
RefSeq |
Entrez ID |
Group A |
Group B |
∆-ß |
Methylation
Fold Change |
Expression
Fold Change |
Adjusted
p value |
Mean |
SD |
Mean |
SD |
FLT4 |
Fms-related tyrosine kinase 4 |
NM_182925.1 |
2324 |
0.10 |
0.06 |
0.64 |
0.19 |
0.53 |
6.13 |
-2.02 |
1.83E-5 |
EPB41L3 |
erythrocyte memb. protein band 4.1L3 |
NM_012307.2 |
23136 |
0.13 |
0.14 |
0.62 |
0.12 |
0.49 |
4.73 |
-2.12 |
3.89E-7 |
FGF4 |
Fibroblast growth factor 4 |
NM_002007.1 |
2249 |
0.08 |
0.06 |
0.57 |
0.24 |
0.48 |
6.9 |
-2.02 |
3.71E-4 |
HTR4 |
5-hydroxytryptamine receptor 4 |
NM_000870.2 |
3360 |
0.08 |
0.04 |
0.55 |
0.15 |
0.46 |
6.6 |
-2.04 |
2.15E-5 |
EDN3 |
Endothelin 3 |
NM_000114.2 |
1908 |
0.08 |
0.06 |
0.52 |
0.20 |
0.44 |
6.79 |
-2.52 |
1.23E-4 |
SLC6A2 |
Solute carrier family 6 member 2 |
NM_001043.2 |
6530 |
0.05 |
0.04 |
0.49 |
0.24 |
0.43 |
9 |
-2.04 |
6.63E-4 |
ZNF134 |
Zinc finger protein 134 |
NM_003435.2 |
7693 |
0.09 |
0.06 |
0.52 |
0.20 |
0.43 |
5.57 |
-2.07 |
1.76E-4 |
GRM6 |
glutamate receptor, metabotropic 6 |
NM_000843.2 |
2916 |
0.12 |
0.06 |
0.54 |
0.23 |
0.42 |
4.6 |
-2.07 |
4.23E-4 |
BCAN |
brevican |
NM_198427.1 |
63827 |
0.10 |
0.03 |
0.51 |
0.23 |
0.41 |
5.23 |
-2.03 |
6.72E-4 |
CDO1 |
Cysteine dioxygenase, type I |
NM_001801.2 |
1036 |
0.05 |
0.05 |
0.45 |
0.21 |
0.40 |
8.9 |
-2.04 |
5.22E-4 |
ZNF671 |
Zinc finger protein 671 |
NM_024833.1 |
79891 |
0.06 |
0.06 |
0.46 |
0.26 |
0.40 |
7.38 |
-2.05 |
0.001 |
PENK |
Proenkephalin |
NM_006211.2 |
5179 |
0.04 |
0.03 |
0.43 |
0.21 |
0.39 |
9.74 |
-2.18 |
6.68E-4 |
EPHA5 |
EPH receptor A5 |
NM_182472.1 |
2044 |
0.10 |
0.06 |
0.49 |
0.19 |
0.39 |
4.7 |
-2.04 |
2.35E-4 |
SLC27A6 |
Solute carrier family 27 |
NM_014031.3 |
28965 |
0.08 |
0.05 |
0.46 |
0.24 |
0.38 |
5.95 |
-2.11 |
0.001 |
COL6A2 |
Collagen type VI alpha 2 |
NM_001849.2 |
1292 |
0.04 |
0.03 |
0.42 |
0.25 |
0.37 |
9.66 |
-2.02 |
0.002 |
AK5 |
Adenylate kinase 5 |
NM_174858.1 |
26289 |
0.07 |
0.05 |
0.44 |
0.20 |
0.37 |
6.57 |
-2.02 |
5.79E-4 |
PTPRN |
Protein tyrosine phosphatase receptor |
NM_002846.2 |
5798 |
0.07 |
0.06 |
0.43 |
0.23 |
0.37 |
6.49 |
-2.04 |
0.002 |
PHOX2A |
Paired-like homeobox 2A |
NM_005169.2 |
401 |
0.07 |
0.06 |
0.42 |
0.23 |
0.36 |
6.27 |
-2.03 |
0.001 |
CDH8 |
Cadherin 8, type 2 |
NM_001796.2 |
1006 |
0.06 |
0.04 |
0.40 |
0.16 |
0.34 |
6.6 |
-2.05 |
2.63E-4 |
GAS7 |
Growth arrest-specific 7 |
NM_003644.2 |
8522 |
0.07 |
0.11 |
0.41 |
0.28 |
0.34 |
5.62 |
-2.18 |
0.007 |
SFRP1 |
secreted frizzled-related protein 1 |
NM_003012.3 |
6422 |
0.09 |
0.07 |
0.44 |
0.27 |
0.34 |
4.63 |
-2.09 |
0.005 |
HYDIN |
hydrocephalus inducing homolog |
NM_017558.2 |
54768 |
0.09 |
0.06 |
0.42 |
0.19 |
0.34 |
4.77 |
-2.02 |
6.49E-4 |
FAM163A |
Family with sequence similarity 163 member A |
NM_173509.2 |
148753 |
0.04 |
0.04 |
0.37 |
0.29 |
0.33 |
9.15 |
-2.07 |
0.008 |
SYNDIG1 |
Synapse differentiation inducing 1 |
NM_024893.1 |
79953 |
0.07 |
0.05 |
0.40 |
0.20 |
0.33 |
5.47 |
-2.04 |
0.001 |
CACNA1G |
Calcium channel voltage-dependent 1G |
NM_018896.3 |
8913 |
0.05 |
0.05 |
0.37 |
0.21 |
0.32 |
7.23 |
-2.07 |
0.002 |
EDNRB |
Endothelin receptor type B |
NT_024524.13 |
1910 |
0.04 |
0.03 |
0.35 |
0.17 |
0.32 |
9.23 |
-2.1 |
5.70E-4 |
CXCL12 |
chemokine (C-X-C motif) ligand 12 |
NM_199168.2 |
6387 |
0.09 |
0.05 |
0.41 |
0.16 |
0.32 |
4.68 |
-2.4 |
3.14E-4 |
RAX |
retina & anterior neural fold homeobox |
NM_013435.1 |
30062 |
0.07 |
0.02 |
0.40 |
0.23 |
0.32 |
5.37 |
-2.04 |
0.003 |
CDX2 |
Caudal type homeobox 2 |
NM_001265.2 |
1045 |
0.06 |
0.03 |
0.37 |
0.14 |
0.30 |
5.89 |
-2.04 |
2.58E-4 |
PRMT8 |
Protein arginine methyltransferase 8 |
NM_019854.3 |
56341 |
0.04 |
0.04 |
0.34 |
0.20 |
0.30 |
8.67 |
-2.03 |
0.002 |
DOK5 |
Docking protein 5 |
NM_018431.3 |
55816 |
0.07 |
0.03 |
0.36 |
0.18 |
0.29 |
5.39 |
-2.05 |
0.001 |
PDGFD |
platelet derived growth factor D |
NM_025208.3 |
80310 |
0.07 |
0.08 |
0.35 |
0.22 |
0.29 |
5.33 |
-2.35 |
0.004 |
ZNF549 |
Zinc finger protein 549 |
NM_153263.1 |
256051 |
0.05 |
0.05 |
0.34 |
0.19 |
0.29 |
6.7 |
-2.03 |
0.002 |
GABRA2 |
Gamma-aminobutyric acid A2 |
NM_000807.1 |
2555 |
0.06 |
0.06 |
0.34 |
0.22 |
0.28 |
5.49 |
-2.03 |
0.006 |
HAND1 |
heart & neural crest derivatives expr.1 |
NM_004821.1 |
9421 |
0.07 |
0.03 |
0.34 |
0.24 |
0.28 |
5.17 |
-2.05 |
0.008 |
STAC |
SH3 and cysteine rich domain |
NM_003149.1 |
6769 |
0.05 |
0.03 |
0.32 |
0.17 |
0.27 |
6.75 |
-2.04 |
0.001 |
GRIK1 |
glutamate receptor ionotropic kainate 1 |
NM_000830.3 |
2897 |
0.06 |
0.04 |
0.32 |
0.20 |
0.26 |
5.24 |
-2.03 |
0.005 |
ADRA2A |
adrenergic alpha-2A-receptor |
NM_000681.2 |
150 |
0.06 |
0.07 |
0.31 |
0.18 |
0.25 |
4.85 |
-2.1 |
0.003 |
COL14A1 |
collagen type XIV alpha 1 |
NM_021110.1 |
7373 |
0.05 |
0.04 |
0.27 |
0.16 |
0.22 |
5.22 |
-2.29 |
0.003 |
MYRIP |
Myosin VIIA and Rab interacting prot. |
NM_015460.2 |
25924 |
0.03 |
0.03 |
0.21 |
0.11 |
0.18 |
6.76 |
-2.03 |
0.002 |
Table 3. Data on selected genes with lower degree of methylation in cervical swabs of CIN III/cervical cancer patients compared with CIN I/CIN II patients and healthy donors. The table shows the 40 genes with the lowest degree of methylation in CIN III and cervical cancer compared to normal cervical tissue.
CpG ID |
Gene |
Description |
RefSeq |
Entrez |
Group A |
Group B |
∆-ß |
Fold
Change |
Adjusted
p value |
Mean |
SD |
Mean |
SD |
cg00400263 |
FAM217B |
family with sequence similarity 217 member B |
NM_022106.1 |
63939 |
0.89 |
0.09 |
0.65 |
0.19 |
-0.24 |
0.73 |
0.006 |
cg17647273 |
UPB1 |
beta-ureidopropionase 1 |
NM_016327.2 |
51733 |
0.86 |
0.07 |
0.63 |
0.16 |
-0.24 |
0.73 |
0.002 |
cg21541083 |
STXBP2 |
syntaxin binding protein 2 |
NM_006949.1 |
6813 |
0.85 |
0.08 |
0.62 |
0.11 |
-0.23 |
0.73 |
2.29E-04 |
cg09830866 |
FAM173A |
family with sequence similarity 173 member A |
NM_023933.1 |
65990 |
0.71 |
0.11 |
0.49 |
0.11 |
-0.22 |
0.68 |
3.54E-04 |
cg25182621 |
CHRD |
chordin |
NM_177978.1 |
8646 |
0.83 |
0.06 |
0.63 |
0.13 |
-0.20 |
0.76 |
0.001 |
cg16869108 |
VHL |
von Hippel-Lindau tumor suppressor |
NT_022517.17 |
7428 |
0.68 |
0.13 |
0.48 |
0.10 |
-0.20 |
0.71 |
0.002 |
cg08124030 |
TM4SF1 |
transmembrane 4 L six family member 1 |
NM_014220.2 |
4071 |
0.71 |
0.13 |
0.53 |
0.15 |
-0.19 |
0.74 |
0.009 |
cg18623836 |
RRM2 |
ribonucleotide reductase regulatory subunit M2 |
NM_001034.1 |
6241 |
0.64 |
0.13 |
0.47 |
0.08 |
-0.18 |
0.73 |
0.003 |
cg18149919 |
PUF60 |
poly(U) binding splicing factor 60 |
NM_014281.3 |
22827 |
0.83 |
0.06 |
0.65 |
0.09 |
-0.17 |
0.79 |
4.04E-04 |
cg13625403 |
TRIM29 |
tripartite motif containing 29 |
NM_058193.1 |
23650 |
0.85 |
0.10 |
0.68 |
0.12 |
-0.17 |
0.80 |
0.004 |
cg02601403 |
TBC1D3C |
TBC1 domain family member 3C |
NM_001001418.2 |
414060 |
0.83 |
0.06 |
0.66 |
0.07 |
-0.17 |
0.80 |
0.007 |
cg02910574 |
CHMP1A |
charged multivesicular body protein 1A |
NM_002768.1 |
5119 |
0.80 |
0.11 |
0.64 |
0.10 |
-0.16 |
0.80 |
0.004 |
cg15046693 |
CEBPG |
CCAAT/enhancer binding protein gamma |
NM_001806.2 |
1054 |
0.57 |
0.08 |
0.41 |
0.11 |
-0.15 |
0.73 |
0.003 |
cg10305797 |
KRTDAP |
keratinocyte differentiation associated protein |
NM_207392.1 |
388533 |
0.58 |
0.14 |
0.43 |
0.07 |
-0.15 |
0.74 |
0.009 |
cg23667432 |
ALPP |
alkaline phosphatase, placental |
NM_001632.2 |
250 |
0.62 |
0.11 |
0.47 |
0.06 |
-0.15 |
0.76 |
0.002 |
cg24691461 |
CCM2L |
CCM2 like scaffolding protein |
NM_080625.2 |
140706 |
0.57 |
0.13 |
0.43 |
0.06 |
-0.14 |
0.75 |
0.009 |
cg26233253 |
SLC35E4 |
solute carrier family 35 member E4 |
NM_001001479.1 |
339665 |
0.69 |
0.10 |
0.55 |
0.09 |
-0.14 |
0.80 |
0.006 |
cg12554573 |
PARP3 |
poly(ADP-ribose) polymerase family member 3 |
NM_001003931.1 |
10039 |
0.59 |
0.09 |
0.46 |
0.09 |
-0.13 |
0.77 |
0.004 |
cg19946699 |
SH3BGR |
SH3 domain binding glutamate rich protein |
NM_007341.2 |
6450 |
0.53 |
0.10 |
0.40 |
0.07 |
-0.13 |
0.75 |
0.003 |
cg21372914 |
CLEC4M |
C-type lectin domain family 4 member M |
NM_014257.3 |
10332 |
0.79 |
0.04 |
0.66 |
0.11 |
-0.13 |
0.83 |
0.006 |
cg02089348 |
TMEM129 |
transmembrane protein 129 |
NM_138385.2 |
92305 |
0.45 |
0.06 |
0.33 |
0.10 |
-0.13 |
0.72 |
0.005 |
cg09547190 |
CARD19 |
caspase recruitment domain family member 19 |
NM_032310.2 |
84270 |
0.52 |
0.08 |
0.39 |
0.06 |
-0.13 |
0.76 |
0.001 |
cg14757296 |
HYAL1 |
hyaluronoglucosaminidase 1 |
NM_007312.3 |
3373 |
0.89 |
0.03 |
0.76 |
0.10 |
-0.12 |
0.86 |
0.007 |
cg17839611 |
GNGT2 |
G protein subunit gamma transducin 2 |
NM_031498.1 |
2793 |
0.71 |
0.10 |
0.59 |
0.05 |
-0.12 |
0.83 |
0.003 |
cg13888886 |
SPO11 |
SPO11, initiator of meiotic double stranded breaks |
NM_012444.2 |
23626 |
0.89 |
0.04 |
0.77 |
0.09 |
-0.12 |
0.86 |
0.004 |
cg03309967 |
PSENEN |
presenilin enhancer gamma-secretase subunit |
NM_172341.1 |
55851 |
0.71 |
0.07 |
0.59 |
0.09 |
-0.12 |
0.83 |
0.005 |
cg26065841 |
CHAC1 |
ChaC glutathione specific gamma-glutamylcyclotransferase 1 |
NM_024111.2 |
79094 |
0.84 |
0.04 |
0.72 |
0.08 |
-0.12 |
0.86 |
0.002 |
cg15952487 |
CD1B |
CD1b molecule |
NM_001764.1 |
910 |
0.52 |
0.09 |
0.41 |
0.07 |
-0.12 |
0.78 |
0.006 |
cg25076881 |
OR10J1 |
olfactory receptor family 10 subfamily J member 1 |
NM_012351.1 |
26476 |
0.46 |
0.11 |
0.34 |
0.05 |
-0.12 |
0.75 |
0.009 |
cg11536940 |
CPQ |
carboxypeptidase Q |
NM_016134.2 |
10404 |
0.24 |
0.09 |
0.13 |
0.05 |
-0.11 |
0.53 |
0.004 |
cg23228178 |
PADI4 |
peptidyl arginine deiminase 4 |
NM_012387.1 |
23569 |
0.53 |
0.08 |
0.42 |
0.07 |
-0.11 |
0.79 |
0.004 |
cg00136477 |
C1QC |
complement C1q C chain |
NM_172369.2 |
714 |
0.70 |
0.07 |
0.59 |
0.07 |
-0.11 |
0.84 |
0.002 |
cg17676574 |
UROC1 |
urocanate hydratase 1 |
NM_144639.1 |
131669 |
0.77 |
0.07 |
0.66 |
0.06 |
-0.11 |
0.86 |
0.001 |
cg00750606 |
CDA |
cytidine deaminase |
NM_001785.1 |
978 |
0.67 |
0.05 |
0.56 |
0.05 |
-0.11 |
0.84 |
3.46E-04 |
cg24387380 |
GABRA5 |
gamma-aminobutyric acid type A receptor alpha5 subunit |
NM_000810.2 |
2558 |
0.58 |
0.10 |
0.47 |
0.06 |
-0.11 |
0.81 |
0.009 |
cg01618851 |
CD209 |
CD209 molecule |
NM_021155.2 |
30835 |
0.54 |
0.07 |
0.43 |
0.06 |
-0.11 |
0.80 |
0.003 |
cg11299964 |
MAPKAP1 |
mitogen-activated protein kinase associated protein 1 |
NM_001006620.1 |
79109 |
0.27 |
0.09 |
0.17 |
0.05 |
-0.11 |
0.61 |
0.006 |
cg06256735 |
MFAP5 |
microfibrillar associated protein 5 |
NM_003480.2 |
8076 |
0.71 |
0.08 |
0.61 |
0.06 |
-0.11 |
0.85 |
0.006 |
cg23988567 |
IGHG3 |
immunoglobulin heavy constant gamma 3 |
NT_026437.13 |
3502 |
0.82 |
0.03 |
0.71 |
0.09 |
-0.11 |
0.87 |
0.009 |
cg26799474 |
CASP8 |
caspase 8 |
NM_001228.3 |
841 |
0.32 |
0.09 |
0.21 |
0.06 |
-0.10 |
0.67 |
0.007 |
Genome-wide promoter methylation analysis
Methylation changes in promoter sequences has often been linked to abnormal transcriptional activity. About half of the known human gene promoters contain CpG islands [22], which are protected from de novo methylation in normal tissues, but often acquire methylation in cancer cells that eventually leads to gene silencing. In order to understand the global changes in methylation that occur during cervical cancer progression and their effects on transcription, we performed a genome-wide DNA methylation profiling of 27578 CpG dinucleotides, located in the promoter region of 14495 genes, using total DNA extracted from cervical swabs of cervical dysplasia (CIN I-CIN III) and cervical cancer patients, as well as healthy donors. The dataset generated can be retrieved from the Gene Expression Omnibus (accession number GSE41384).
To capture the intrinsic nature of the data, statistical analyses were performed by means of unsupervised hierarchical clustering. We decided to exclude one CIN I sample, because it presented a markedly different behavior with respect to the rest of samples. Unsupervised analysis (Hierarchical Clustering) uncovered that the samples tend to cluster into two groups based on the similarity (measured in Euclidian distances) of methylation profiles: Group A, comprising samples negative for intraepithelial lesion, as well as CIN I and CIN II samples; and Group B, including CIN III, carcinoma in situ and invasive cancer samples. In a second step, supervised T-Test analysis was performed to identify genes with significant differences in their methylation levels between the groups detected in the unsupervised analysis. This analysis showed that, during cervical cancer progression, hypermethylation of CpG islands within gene promoters is significantly more frequent than hypomethylation (Figure 1A). Thus, CpG islands in the promoters of 1069 genes were hypermethylated in the group B, while a remarkably lower number of genes (85) were hypomethylated in this group (p<0.0001) (Figure 1A and Supplementary table 4). The heatmap in figure 1A illustrates the differential methylation of gene promoters in healthy/CIN I/CIN II samples compared with those in CIN III/cervical cancer samples. The complete list of the 1069-hypermethylated genes arranged by their Ƨ-values is shown in the supplementary table 4 together with their fold-change and the p-values for the Student t-test applied to the statistic analysis of the data.
The table 1 displays a list of the 40 genes with the highest ∆ß-values in CIN III and cervical cancer (group B). The criteria for selection of these genes were: (i) a mean basal ß-value < 0.2 in group A (healthy/CIN I/CIN II samples) and (ii) a ∆ß-value > 0.45 (calculated by subtracting the mean ß-values in the group A from those in group B). The three top genes in this list are IGSF21, UTF1 and PTGDR with ∆ß-values of 0.59, 0.57 and 0.55, respectively. This approach also revealed six genes with a fold change in methylation > 10: UTF1, CADPS, ELMO1, ZNF132, NELL1 and SLC18A3.
In order to compare and integrate the genes differentially methylated in our study population with gene expression profiles described previously in cervical cancer samples, we selected and analyzed a number of gene expression microarrays of public access [19]. The microarray platform used was the Affymetrix HG-U133A in the Gene Expression Omnibus (GEO, www.ncbi.nlm.nih.gov/geo/) accession number GSE780319. The selection of differentially expressed genes was performed with an unsupervised cluster analysis with Euclidian distance. The analysis was performed using the same software as for the methylation microarrays to compare gene expression in cervical cancer and normal cervical samples and applying the same cut-off for statistical significance (p≤0.0001).
The figure 1B and C shows the result of such analysis, by which 1726 genes were found down-regulated in cervical cancer compared with normal cervical tissue. When these genes were confronted with the 1069 with hypermethylated promoters, we identified a set of 132 genes (12.3%) common to both groups (Figure 1C, Supplementary table 5). The heatmap in figure 1B shows a supervised hierarchical clustering of the hypermethylated genes whose expression was lower in cervical cancer compared to normal cervical tissue. The table 2 contains data of the 40 genes with the highest degree of promoter methylation (Ƨ-values between 0.53 and 0.18, fold change ranging between 9.74 and 4.06) and with the lowest expression level (fold change between -2.18 and -2.07) in CIN III and cervical cancer compared to normal cervical tissue. The top three genes in this list are: FLT4 (Fms-related tyrosine kinase 4), EPB41L3 (erythrocyte membrane protein band 4.1L3), FGF4 (Fibroblast growth factor 4), with Ƨ-values of 0.53, 0.49 and 0.48, respectively.
As shown in the figure 1 our analysis uncovered also 85 hypomethylated genes (Supplementary table 6). The Table 3 contains the data of a selection of 40 genes with lower degree of promoter methylation in CIN III and cervical cancer and with the highest -Ƨ-values (-0.24 to -0.10). There are six genes with a -Ƨ of -0.20 or above: FAM217B (family with sequence similarity 217 member B), of unknown function, UPB1 (beta-ureidopropionase 1), STXBP2 (syntaxin binding protein 2), FAM173A, CHRD (chordin) and VHL (von Hippel-Lindau tumor suppressor). Furthermore, our supervised T-test analysis revealed 1945 genes with augmented expression in cervical cancer compared to normal tissue. In contrast to the hypermethylated genes, contingency tables showed that only as few as 4 out of the 85-hypomethylated genes (4.7%) were in the group of genes with increased expression in cervical cancer: NCAP-G (a subunit of the condensin complex), PSENEN (presenilin gamma-secretase subunit), HIST1H3H (Histone cluster 1 H3 family member H) and RRM2 (Ribonucleotide reductase regulatory subunit M2) with -Ƨ-values of -0.05, -0.12, -0.07 and -0.18, respectively (Table 3 and supplementary table 6).
Differentially methylated functional gene clusters in neoplastic cervical lesions
Gene ontology analysis of CpG islands at promoter regions displaying significantly higher methylation profiles in advanced cervical lesions and cervical cancer compared to low-grade lesions and normal tissue revealed several gene clusters.
We used the bioinformatics tool Database for Annotation, Visualization and Integrated Discovery (DAVID) [17] (URL david.abcc.ncifcrf.gov) for the analysis of genes based on Gene Ontology (GO). To analyze the hyper and hypomethylated genes whose heat map is shown in figure 1, we applied the Functional Annotation Tool of DAVID, which provides information on biological functions enriched in a gene list relative to all annotated genes.
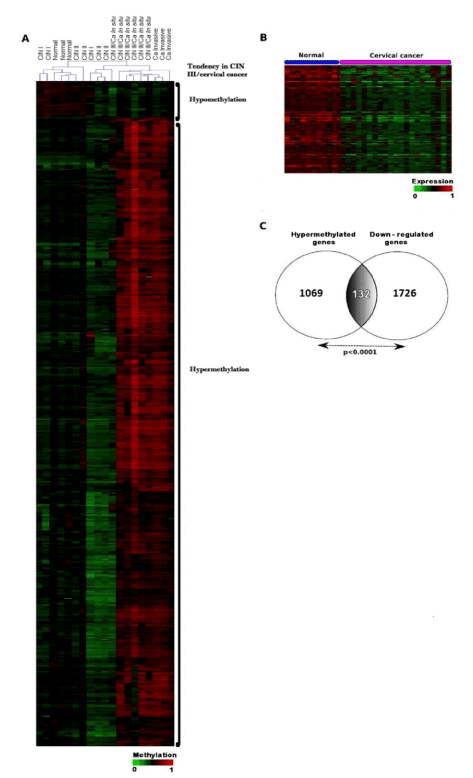
Figure 1. Clustering analysis of methylation and correlation between hypermethylation and gene expression in a subset of 132 genes. A. Supervised hierarchical clustering of gene promoter methylation in cervical samples from CIN, carcinoma in situ and invasive cancer patients as well as healthy donors. Differentially methylated genes were identified by a supervised T-Test analysis in which only those with a p≤0.0001 were selected. The heatmap of the methylation values discriminates between two subsets of cervical samples: normal and low-grade lesions (CIN I and CIN II), on the left side, and CIN III/cancer (in situ and invasive) on the right side of the panel. The intensity of the colors represents the gradient of the Δβ-values, ranging from 0 (bright green) to 1 (bright red). The results suggest that the promoter regions of these genes are sites of increased methylation heterogeneity in advanced cervical dysplasia and cancer. B. Heatmap from a supervised hierarchical clustering of 132 hypermethylated genes with down-regulated expression in cervical cancer compared to normal tissue. Patients from GSE7803 were included. A T-test analysis with p=0.0001 as cut-off was used. The intensity of the colors represents the gradient of expression ranging from 0 (bright green) to 1 (bright red). C. Venn Diagram of contingency showing an overlapping group of hypermethylated and down-regulated genes in CIN III and cervical cancer (p<0.0001).
The left panel in figure 2 (A and B) shows the GO-analysis of the 132 genes with higher degree of promoter methylation in CIN III / cervical cancer irrespective of the gene expression data, These genes were selected as the first 132 genes in the list of hypermethylated genes arranged by their Ƨ-values (Supplementary table 4). The results, after applying REVIGO [18] to eliminate redundant terms, showed a higher frequency of transcription and cell structure genes (27% and 26%, respectively), followed by genes involved in neuronal activity, signal transduction, cell cycle and metabolism (14%, 12% 8% and 8%, respectively). An interactive graph of biological processes based on the GO analysis is shown in figure 2 B.
The right panel in figure 2 (C and D) shows the GO-analysis of the 132 genes hypermethylated and down-regulated in CIN III / cervical cancer, after applying REVIGO [18] to eliminate redundant terms, showed a prevalence of genes involved in cell structure (27.4%), followed by genes related to transcription (24.3%), metabolism (10%), neuronal function (6.8%), cell cycle regulation (5.7%), development (4.1%) and other cellular processes such as regulation of T cell activation, ion transport and cell adhesion (Figure 2 C and D). The figure 2 C shows an Interactive graph of biological processes based on the ontology analysis and the gene clusters of the 132 hypermethylated and down-regulated genes. The nodes with higher gene representation are related to proliferation, signaling, adhesion and cell-cell communication.
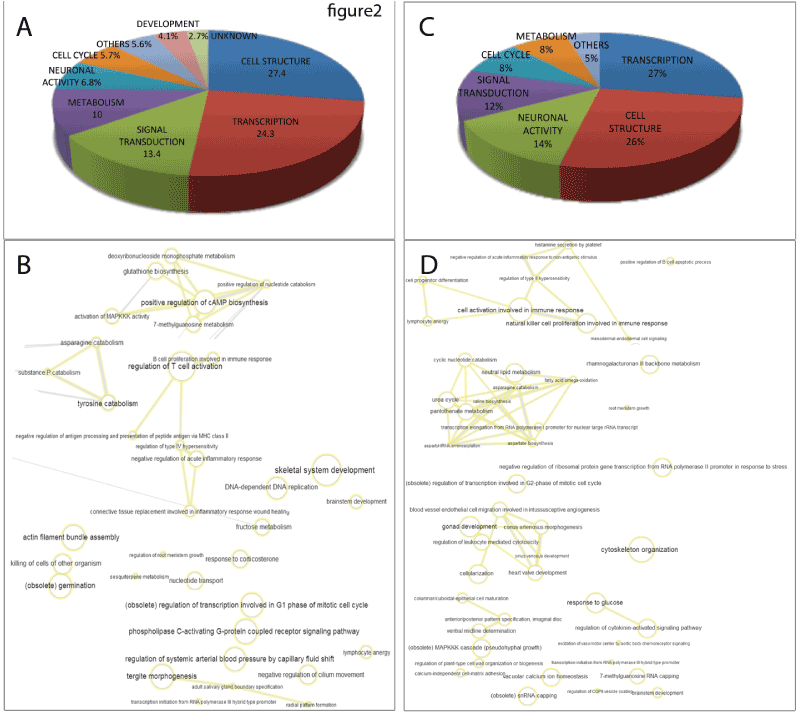
Figure 2. Gene ontology (GO) enrichment analysis of the genes with higher methylation in CIN III and cervical cancer compared with CIN I/CIN II and healthy donor samples. A and B show the analysis of 132 genes with hypermethylated promoter region in CIN III and cervical cancer. C and D show the analysis of genes with hypermethylated promoter region associated with diminished expression in CIN III and cervical cancer. A and C: Fraction distribution generated with Panther of GO terms based on molecular function, biological process and cellular component. B and D: Analysis in GO terms plotted following REVIGO protocols resulting in an interactive graph of biological process based on the ontology analysis and the gene clusters of the hypermethylated and down-regulated genes. The size and color intensity of the nodes are proportional to the number of genes associated with the specific term and the statistical significance (p value), respectively. The thickness of the lines joining the nodes is proportional to the proximity of the regulatory or biochemical pathways in which they interact.
The figure 3 shows the GO enrichment analysis of the 85 genes with a lower degree of methylation in CIN III / cervical cancer. The fraction distribution of these genes (Figure 3 A) shows a higher incidence of genes related to cell structure (22%) and metabolism (13%), but significantly less genes involved in transcription regulation (8%) than in the group of hypermethylated genes. Cell cycle and development genes represent 6% each. In addition, a large proportion of genes (34%) are involved in a variety of functions, such as apoptosis, organelle organization and secretion, ion and sugar transport and regulation of immune responses. The four hypomethylated genes with enhanced expression in CIN III / cervical cancer are basically involved in regulation of gene expression and signal transduction.
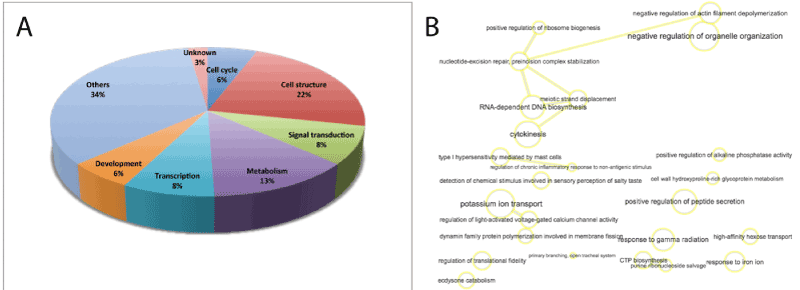
Figure 3. GO enrichment analysis of the 85 hypomethylated genes in CIN III and cervical cancer. A. Fraction distribution generated with Panther of hypomethylated genes GO terms based on molecular function, biological process and cellular component. B. Analysis in GO terms plotted following REVIGO protocols. Interactive graph of biological process based on the ontology analysis and the gene clusters of the hypomethylated genes. The size of the nodes are proportional to the number of genes associated with the specific term and the statistical significance (p value), respectively. The thickness of the lines joining the nodes is proportional to the proximity of the regulatory or biochemical pathways in which they interact.
Genes with apparent correlation between promoter methylation status and gene expression
An interesting group of genes that were hypermethylated in our study, and whose expression was previously found diminished in cervical cancer, belonged to the cadherin family. Thus, we found hypermethylated genes encoding cell adhesion proteins, such as PCDH11X, CDH22, CDH12 and CDH8. Previous methylation studies using MeDIP-chip analysis showed that the promoter of PCDH11X was hypermethylated in CIN III [23]. Also the promoter of CDH22 has been reported hypermethylated in breast cancer tissue compared to adjacent normal tissue. In addition, hypermethylation of CDH22 correlated with poor prognosis regardless of age and tumor stage [24]. Our results confirm these data and extend them to increased methylation of cadherin CDH12 in CIN III and cervical cancer. CDH12 is a large gene located in chromosome 5p14.3 close to FRA5E, a region of genomic instability, which belongs to a group of about 90 common fragile sites (CFS) distributed throughout the human genome, where oncogenic HPVs frequently integrate [22]. Hypermethylation of CDH12 might be related to HPV integration, although this event remains to be tested in our samples. In agreement with our data, a recent prospective cross-sectional study, showed promoter hypermethylation of cadherin 8, type 2 (CDH8) and another two tumor-suppressor candidate genes (adenylate cyclase 8, ADCY8, and zinc finger protein 582, ZNP528), as quantified by bisulfite-pyrosequencing of genomic DNA. Hypermethylation of these genes correlated with worsening grade [26].
The gene EDN3 (endothelin 3), encoding a vasoactive peptide, was hypermethylated in CIN III/cancer samples. This gene has been recently reported hypermethylated and down-regulated in CIN III lesions suggesting its involvement in the pathogenesis of cervical cancer [27,28]. Interestingly, the gene encoding the endothelin receptor type B (EDNRB) was also hypermethylated in CIN and cancer tissues in a recent study [29].
We also identified members of the SFRP family hypermethylated in CIN III and cervical cancer, in particular SFRP1 (Secreted Frizzled Related Protein 1), which acts as modulator of Wnt signaling through direct interaction with Wnt family members. Epigenetic silencing of SFRP genes leads to deregulated activation of the Wnt-pathway, which is associated with cancer. SFRP1 was previously found hypermethylated and silenced in invasive cervical cancer [30] and cervical cancer-derived cell lines [31].
The TP73 gene is homologous to p53 gene, involved in cell cycle control in response to DNA damage, TP73 was hypermethylated in CIN III and cancer in the present study, correlating with lower expression in cervical cancer, similar to results reported previously [32,33].
The AR (androgen receptor) gene, which has been reported underexpressed in CIN III/cervical cancer compared to CIN I/CIN II [34], was differentially methylated in these two groups with increasing methylation during progression from CIN I to cancer.
Some genes related to the immune system, such as CXCL12 and IL1b, were hypermethylated in CIN III and cervical cancer. Recently, CXCL12 has been shown hypermethylated in cervical cancer biopsies and in established cell lines (HeLa, SiHa) and its expression down regulated [35].
There is an imperative demand for the identification of markers that enable predict progression of cervical intraepithelial neoplasia to invasive cancer, thus providing better support for decisions on the follow up and handling of this disease, especially at the pre-cancer stages. Biomarkers associated with DNA methylation are revealing promising11. DNA methylation is a normal cellular process used to silence genes and regulate gene dosage. Yet aberrations in DNA methylation, most notably in gene promoter regions, have been observed in the process of carcinogenesis from tumor initiation to metastasis [36]. Indeed, differential methylation and, in particular, hypermethylation has been frequently described in many types of tumors with respect to the tissues from which they originate [37].
This study was aimed at identifying gene promoter regions differentially methylated in samples of patients with either advanced precursor intraepithelial lesions or cervical cancer as compared to samples of healthy donors with no evidence of cervical pathology. In addition, we intended to determine whether the changes in gene promoter methylation were correlated with differential expression of the corresponding encoded proteins during cervical cancer progression.
2021 Copyright OAT. All rights reserv
Previous studies evaluating cervical cancer epigenetics have shown differential methylation of the promoter region of specific genes in normal and cervical cancer tissues [30]. Indeed, aberrant methylation has been described from the very early stages of cervical carcinogenesis. Besides their mechanistic implications, the genes that are differentially methylated in tumor cells may serve as biomarkers for early detection of the disease as well as be of prognostic value [38]. However, studies available to date reveal somehow inconclusive and, in some cases, conflicting results [10]. This could be explained by the different analytical techniques employed, the ethnic/genetic variability of the populations under study, differences in the HPV types present in the samples, and also the diversity of genes evaluated in such studies [39]. Some of these genes were CDH1 (E-cadherin), DAPK1 (death-associated protein kinase 1), CADM1 (cell adhesion molecule 1), RARB and TERT (telomerase). In general, these studies described 29 to 33% methylation in samples with high-grade cervical lesions (CIN II / CIN III) related with progression to cervical cancer, without reaching a conclusion on the possible predictive potential in cervical cancer progression [10].
It has become evident, however, that only a wide simultaneous analysis of the genome may allow identify subsets of differentially methylated genes associated with the development of cervical cancer, which could be of high prognostic significance. These studies should provide new data on patterns of DNA methylation associated with cervical cancer progression, thereby increasing the number of candidate genes that could serve as prognostic biomarkers. In a previous study analyzing the genome-wide methylation pattern of high-grade (CIN III) and healthy women, COL25A1 and KATNAL2 were identified as hypermethylated and were suggested as candidate biomarkers for early detection of cervical cancer [23]. Furthermore, in a more recent study, Farkas et al. [38] described a subset of 24 differentially methylated genes in cervical cancer compared with CIN3 and normal cervical tissue.
In the present study we used the Illumina platform to evaluate the methylation profiles of the genomes of cervical swabs of patients at various stages of cervical carcinogenesis (CIN I to CIN III and cervical cancer) and compared them with those of healthy donors. The results were examined applying an unsupervised cluster analysis which allowed identify two distinct methylation profiles, one corresponding to healthy donor, CIN I and CIN II (group A) and another to CIN III and cervical cancer (group B). Due to the limited number of samples, it was not possible to assess a statistically significant relationship between the HPV type (and copy number) and the methylation status of individual genes within the two groups.
Hypermethylation frequently leads to down-regulation of gene expression. DNA-binding proteins containing methyl-CpG binding domains or C2H2 zinc fingers bind to methylated DNA and mediate transcriptional repression of methylated promoters [40]. Therefore, we considered essential to compare the genes that we found to have hypermethylated promoters in the group CIN III/cervical cancer patients with the expression profile of these genes in a gene expression analysis performed in a comparable study population using a microarray platform (Affymetrix HG-U133A). In this cross-analysis we found that, of the 1069 hypermethylated genes that we identified, only the expression of 120 genes (~11%) was down-regulated in the Affymetrix HG-U133A gene expression array, while the expression of the remainder hypermethylated genes did not change (Figure 1 and table 2).
The majority of hypermethylated and down-regulated genes identified in the present study are involved in cellular transcription and transduction pathways, only a few are involved in metabolism and cell adhesion. From the known functions of these genes it can be inferred that their repression would facilitate the process of carcinogenesis [41]. The analysis of the genes found differentially hypomethylated in the present study pinpointed four genes that have been found overexpressed in CC: HCAP-G, PSEN, HIST1H3H and RRM2. Increased transcription of the genes AKR1B10 genes, TPX2, HCAP-G and RRM2 has been described previously in hepatocellular carcinoma [42]. In addition, it has been shown that E7 increases the expression of RRM2 in tumor tissue as well as in cervical cancer cell lines [43]. They were suggested as potential therapeutic targets, since their inhibition by interference RNA decreased proliferation of the tumor cells in vitro. Recently, the expression of PSEN1, a subunit of the gamma-secretase complex, has been found increased in locally advanced cervical cancer compared to normal cervical tissue [44]. As for histone cluster 1 H3h, HIST1H3H, it is known to be overexpressed in cervical squamous cancer tissue compared to normal tissue (Human Protein Atlas available from www.proteinatlas.org). Moreover, a recent study combining transcriptomics, tissue microarray and molecular docking showed enhanced expression of this gene in renal cell carcinoma [45].
In conclusion there is a trend indicating that the methylation profile is similar in cervical swab samples from patients with CIN III and cervical cancer and clearly distinct of samples from with CIN I, CIN II patients and subjects with negative pathology. Further studies with larger numbers of patients should determine which of the genes identified here as differentially methylated have greater prognostic value.
The authors have no conflict of interest.
Universidad Nacional de Colombia, Colciencias (number 1101-519-28973) and German Cancer Research Center (DKFZ)
We are grateful to those women who donating their samples made possible this study. We thank Prof. Harald zur Hausen for continuous support and discussions and Victoria Juarez for critical reading of the manuscript. This work was partly supported by a grant of COLCIENCIAS (110151928973, 519-2010) to AGR and FAA.We are grateful to those women who donating their samples made possible this study. We thank Prof. Harald zur Hausen for continuous support and discussions and Victoria Juarez for critical reading of the manuscript. This work was partly supported by a grant of COLCIENCIAS (110151928973, 519-2010) to AGR and FAA.
View supplementary data
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, et al. (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127: 2893-2917. [Crossref]
- zur Hausen H1 (2009) Papillomaviruses in the causation of human cancers - a brief historical account. Virology 384: 260-265. [Crossref]
- Muñoz N, Bosch FX, de Sanjosé S, Herrero R, Castellsagué X, et al. (2003) Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med 348: 518-527. [Crossref]
- Wise-Draper TM, Wells SI (2008) Papillomavirus E6 and E7 proteins and their cellular targets. Front Biosci 13: 1003-1017. [Crossref]
- Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S (2007) Human papillomavirus and cervical cancer. Lancet 370: 890-907. [Crossref]
- Shen H, Laird PW (2013) Interplay between the cancer genome and epigenome. Cell 153: 38-55. [Crossref]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, et al. (2009) The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41: 178-86. [Crossref]
- Bibikova M, Fan JB (2010) Genome-wide DNA methylation profiling. Wiley Interdiscip Rev Syst Biol Med 2: 210-223. [Crossref]
- Fernandez AF, Rosales C, Lopez-Nieva P, Grana O, Ballestar E, et al. (2009) The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res 19: 438-51. [Crossref]
- Wentzensen N, Sherman ME, Schiffman M, Wang SS (2009) Utility of methylation markers in cervical cancer early detection: appraisal of the state-of-the-science. Gynecol Oncol 112: 293-9. [Crossref]
- Shivapurkar N, Sherman ME, Stastny V, Echebiri C, Rader JS, et al. (2007) Evaluation of candidate methylation markers to detect cervical neoplasia. Gynecol Oncol 107: 549-53. [Crossref]
- Schmitt M, Dondog B, Waterboer T, Pawlita M (2008) Homogeneous amplification of genital human alpha papillomaviruses by PCR using novel broad-spectrum GP5+ and GP6+ primers. J Clin Microbiol 46: 1050-9.
- Garcia DA, Cid-Arregui A, Schmitt M, Castillo M, Briceno I, et al. (2011) Highly Sensitive Detection and Genotyping of HPV by PCR Multiplex and Luminex Technology in a Cohort of Colombian Women with Abnormal Cytology. Open Virol J 5: 70-9. [Crossref]
- Schmitt M, Bravo IG, Snijders PJ, Gissmann L, Pawlita M, et al. (2006) Bead-based multiplex genotyping of human papillomaviruses. J Clin Microbiol 44: 504-512. [Crossref]
- Chu VT, Gottardo R, Raftery AE, Bumgarner RE, Yeung KY (2008) MeV+R: using MeV as a graphical user interface for Bioconductor applications in microarray analysis. Genome Biol 9: R118. [Crossref]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I (2001) Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125: 279-284. [Crossref]
- Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44-57. [Crossref]
- Supek F, Bošnjak M, Škunca N, Šmuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6: e21800. [Crossref]
- Zhai Y, Kuick R, Nan B, Ota I, Weiss SJ, et al. (2007) Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res 67: 10163-72.
- Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, et al. (2011) Increased methylation variation in epigenetic domains across cancer types. Nat Genet 43: 768-75.
- Teschendorff AE, Widschwendter M (2012) Differential variability improves the identification of cancer risk markers in DNA methylation studies profiling precursor cancer lesions. Bioinformatics 28: 1487-94.
- Larsen F, Gundersen G, Lopez R, Prydz H (1992) CpG islands as gene markers in the human genome. Genomics 13: 1095-1107. [Crossref]
- Lendvai A, Johannes F, Grimm C, Eijsink JJ, Wardenaar R, et al. (2012) Genome-wide methylation profiling identifies hypermethylated biomarkers in high-grade cervical intraepithelial neoplasia. Epigenetics 7: 1268-78. [Crossref]
- Martin-Sanchez E, Mendaza S, Ulazia-Garmendia A, Monreal-Santesteban I, Cordoba A, et al. (2017) CDH22 hypermethylation is an independent prognostic biomarker in breast cancer. Clin epigenetics 9: 7. [Crossref]
- Gao G, Smith DI (2016) Role of the Common Fragile Sites in Cancers with a Human Papillomavirus Etiology. Cytogenetic and genome research 2016;150: 217-26. [Crossref]
- Shen-Gunther J, Wang CM, Poage GM, Lin CL, Perez L, et al. (2016) Molecular Pap smear: HPV genotype and DNA methylation of ADCY8, CDH8, and ZNF582 as an integrated biomarker for high-grade cervical cytology. Clinical epigenetics 8: 96. [Crossref]
- Chen YC, Huang RL, Huang YK, Liao YP, Su PH, et al. (2014) Methylomics analysis identifies epigenetically silenced genes and implies an activation of beta-catenin signaling in cervical cancer. Int J Cancer 135: 117-27. [Crossref]
- Liu MY, Zhang H, Hu YJ, Chen YW, Zhao XN (2016) Identification of key genes associated with cervical cancer by comprehensive analysis of transcriptome microarray and methylation microarray. Oncology letters 12: 473-8.
- Vasiljevic N, Scibior-Bentkowska D, Brentnall AR, Cuzick J, Lorincz AT (2014) Credentialing of DNA methylation assays for human genes as diagnostic biomarkers of cervical intraepithelial neoplasia in high-risk HPV positive women. Gynecol Oncol 132: 709-14. [Crossref]
- Sova P, Feng Q, Geiss G, Wood T, Strauss R, et al. (2006) Discovery of novel methylation biomarkers in cervical carcinoma by global demethylation and microarray analysis. Cancer Epidemiol Biomarkers Prev 15: 114-23. [Crossref]
- Chung MT, Sytwu HK, Yan MD, Shih YL, Chang CC, et al. (2009) Promoter methylation of SFRPs gene family in cervical cancer. Gynecol Oncol 112: 301-306. [Crossref]
- Craveiro R, Costa S, Pinto D, Salgado L, Carvalho L, et al. (2004) TP73 alterations in cervical carcinoma. Cancer Genet Cytogenet 150: 116-21. [Crossref]
- Henken FE, Wilting SM, Overmeer RM, van Rietschoten JG, Nygren AO, et al. (2007) Sequential gene promoter methylation during HPV-induced cervical carcinogenesis. Br J Cancer 97: 1457-64. [Crossref]
- den Boon JA, Pyeon D, Wang SS, Horswill M, Schiffman M, et al. (2015) Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc Natl Acad Sci U S A 112: E3255-64. [Crossref]
- Yadav SS, Prasad SB, Prasad CB, Pandey LK, Pradhan S, et al. (2016) CXCL12 is a key regulator in tumor microenvironment of cervical cancer: an in vitro study. Clin Exp Metastasis 33: 431-9. [Crossref]
- Shukla HD, Mahmood J, Vujaskovic Z (2015) Integrated proteo-genomic approach for early diagnosis and prognosis of cancer. Cancer lett 369: 28-36. [Crossref]
- Sproul D, Kitchen RR, Nestor CE, Dixon JM, Sims AH, et al. (2012) Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns. Genome Biol 13: R84. [Crossref]
- Farkas SA, Milutin-Gasperov N, Grce M, Nilsson TK (2013) Genome-wide DNA methylation assay reveals novel candidate biomarker genes in cervical cancer. Epigenetics 8: 1213-25. [Crossref]
- Nephew KP (2009) What will it take to obtain DNA methylation markers for early cervical cancer detection? Gynecol Oncol 112: 291-2.
- McCabe MT, Brandes JC, Vertino PM (2009) Cancer DNA methylation: molecular mechanisms and clinical implications. Clin Cancer Res 15: 3927-3937. [Crossref]
- Kalari S, Pfeifer GP (2010) Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv Genet 70: 277-308. [Crossref]
- Satow R, Shitashige M, Kanai Y, Takeshita F, Ojima H, et al. (2016) Combined functional genome survey of therapeutic targets for hepatocellular carcinoma. Clin Cancer Res 16: 2518-28. [Crossref]
- Wang N, Zhan T, Ke T, Huang X, Ke D, et al. (2014) Increased expression of RRM2 by human papillomavirus E7 oncoprotein promotes angiogenesis in cervical cancer. Br J Cancer 110: 1034-44. [Crossref]
- Campos-Parra AD, Padua-Bracho A, Pedroza-Torres A, Figueroa-Gonzalez G, Fernandez-Retana J, et al. (2016) Comprehensive transcriptome analysis identifies pathways with therapeutic potential in locally advanced cervical cancer. Gynecol Oncol 143: 406-13. [Crossref]
- Karim S, Al-Maghrabi JA, Farsi HM, Al-Sayyad AJ, Schulten HJ, et al. (2016) Cyclin D1 as a therapeutic target of renal cell carcinoma- a combined transcriptomics, tissue microarray and molecular docking study from the Kingdom of Saudi Arabia. BMC cancer 16: 741. [Crossref]