Abstract
Dilated cardiomyopathy is the most prevalent phenotype. It is a frequent cause of heart failure and the primary indication for heart transplantation. Although it has specific standardized criteria for diagnosis, the criteria establish DCM as a non-specific phenotype caused by genetic factors, environmental factors or a combination of both. The diagnosis consists of a wide range of other cardiomyopathies that lead to a dilated ventricular wall, which has made differential diagnosis challenging and many studies providing varying findings. The lack of specific clinical management guidelines has influenced the use of general management concepts of LV dysfunction and heart failure, which vary between studies. This paper reviews current evidence on DCM to provide a comprehensive understanding of its clinical manifestations, etiology, diagnosis and clinical management.
Key words
dilated cardiomyopathy, heart failure, non-ischemic cardiomyopathy
Introduction
Congestive heart failure (CHF) due to poor cardiac function is a serious heart condition and the leading cause of morbidity and mortality in pediatric, adult and geriatric populations worldwide. The disorder is also the most common indication for cardiac transplantation with annual cost implications in the U.S. alone approximated at $200 million [1]. Effective management of CHF is thus critical to improved clinical management, reduced associated cost of healthcare and improved patient survival. One of the major focus on clinical management of CHF is the prevention and management of its main etiologies. In this approach, cardiomyopathies have received extensive attention. Cardiomyopathies define a heterogeneous group of disorders of the myocardium (cardiac musculature) characterized by cardiomyocytes injury and necrosis, myocardial tissue fibrosis, and associated mechanical and electrical cardiac dysfunction [2]. The World Health Organization (WHO)/International Society and Federation of Cardiology (ISFC) Task Force classifies cardiomyopathies into four major morphological and functional phenotypes: (a) dilated cardiomyopathy (DCM); (b) hypertrophic cardiomyopathy (HCM); (c) restrictive cardiomyopathy (RCM); and (d) arrhythmogenic right ventricular dysplasia (ARVD) cardiomyopathy [3]. Other studies classify cardiomyopathies as primary (due to genetic/familial etiologies) or acquired (due to exposure to environmental factors). In either classification, DCM is the most frequently diagnosed phenotype [4]. Thus, the precise knowledge of risk factors, clinical manifestation, prognosis, diagnosis and clinical management of DCM would contribute considerably to improvement in the management of CHF.
Clinical Description
Definition
The 1995 WHO/ISFC Task Force on the Definition and Classification of Cardiomyopathies initially defined DCM as a spectrum of heterogeneous myocardial disorders characterized by ventricular dilation and depressed myocardial function in the absence of hypertension, valvular, congenital or ischemic heart disease [3]. In 2016, the European Society of Cardiology (ESC) working group on myocardial and pericardial diseases proposed the most recent and widely referenced clinicopathologic definition of DCM. It is a progressive and usually irreversible myocardial disorder characterized by left ventricular (LV) or biventricular (BV) dilation and global systolic (contractile) dysfunction not explained by chronic abnormal loading conditions such as hypertension and valvular disorders or coronary artery disease (CAD) sufficient to cause global systolic impairment [5]. The Italian Federation of Cardiology [6] defines DCM as a primary myocardial disease characterized by dilation and systolic dysfunction of the left or both ventricles in the absence of abnormal hemodynamic overload or CAD in sufficient amounts to cause ventricular dilation or the hypokinetic left ventricle.
From the definitions, DCM causes the cardiac muscles to dilate (lengthen and thin) leading to significant deterioration of LV contractile function, ventricular and supraventricular arrhythmias, abnormalities in cardiac conduction system, thromboembolism and a frequent culmination in heart failure in the later stages of the disorder [3] [7]. However, in clinical practice, the pathogenesis of CHF falls into two categories: (a) ischemic and (b) non-ischemic cardiomyopathy where the latter has been interchangeably used with DCM. Although practical, non-ischemic cardiomyopathy may include cardiomyopathies due to hemodynamic (volume or pressure) overload such as hypertension and valvular heart disease that are not part of the accepted definition of DCM. Ischemic cardiomyopathy, on the other hand, defines a type of cardiomyopathy secondary to ischemic heart disease or myocardial infarction in sufficient amounts to cause LV dysfunction [8].
Classification
Although professional heart/cardiologists associations recognize DCM as a distinct disease entity, it is indeed the final common pathway for a wide variety of different etiologic mechanisms. These mechanisms are not always clear and implicated frequently as the underlying reason for the discrepancies in the current international classification of DCM [6]. The American Heart Association (AHA) classifies DCM as a primary cardiomyopathy when the condition affects only the myocardium and secondary cardiomyopathy when it is a part of a multi-organ disease [8]. The European Society of Cardiology (ESC) classifies DCM into genetic familial form and non-genetic familial form [5]. Such differences in etiologic classification of DCM from leading professional cardiology or heart associations has important clinical implications in diagnostic, therapeutic and prognostication of DCM. A key implication is that diagnosis should consider all the potentially reversible underlying causes of LV dysfunction, which could be potentially reversed by the administration of specific therapeutic interventions [8].
Epidemiology
Status of Epidemiology
Epidemiology provides statistical estimates of the prevalence, incidence and distribution of health and disease conditions in defined populations [9]. It is an important component of public health helpful for shaping policy decisions on disease management and guiding evidence-based studies [10]. However, establishing the exact epidemiology of DCM has been problematic and thus its prevalence and incidence have remained poorly defined [11]. This is because diagnosis of DCM lacks strict clinical guidelines; its subclinical phase is asymptomatic and usually missed, and diagnosis is often always incidental. Moreover, DCM has low frequency in the general population; and more importantly, most reports establish diagnosis using autopsy analysis [2]. Current studies have also not specifically examined population-based epidemiology of DCM [12]. Thus, available estimates of its prevalence and incidence rates are not recent and in addition use older diagnostic modalities that have a high likelihood of underestimating the current disease prevalence [7].
Prevalence and Incidence
However, the latest available research-based estimates provide a wide range of prevalence and incidence of DCM. A widely quoted incidence rate of DCM is an old U.S. study [13], which estimated an incidence rate of between 3.9 and 7.9 per 100,000 person-years between the first and fifth year of the study. In Europe, the estimated autopsy incidence rate of DCM is 4.5 cases in 100,000 people per year and the clinical incidence is 2.45 cases per 100,000 people translating into 6.95 incidence per 100,000 people per year [11]. A prevalence of up to 36 cases per 100,000 have also been reported [2]. The wide differences in the reported incidence rates are the result of geographic variation, patient selection and diagnostic criteria adopted in individual studies [13].
DCM clinically manifests at all ages but it is more common in individuals in their thirties and forties. Thus, it is more prevalent in adults, affecting about 7.5 in 100,000 compared to children and teenagers (<18years) affecting 0.57 in 100,000 [14]. Furthermore, the incidence of DCM varies by gender, race and age. In adults, it is more common in males than in females [15]. In children (<18 years), the annual incidence rate in 100,000 is higher in boys than in girls (0.66 vs. 0.47), higher in blacks than in whites (0.98 vs. 0.46) and higher in infants than in children (4.40 vs. 0.34) [14]. These are older estimates, creating the need for population-based epidemiology studies on DCM using newer diagnostic modalities to establish an up-to-date prevalence and incidence rate both globally, and in different geographic areas as well as in different patient populations [7].
Clinical Manifestation
Signs and Symptoms
DCM first presents with signs and symptoms of heart failure (HF) due to high cavitary blood volume or low cardiac output. At diagnosis, the most frequent symptoms are severe impairment of LV ejection function and patients’ classification into New York Heart Association (NYHA) functional class III to IV. About 20-35% experience angina-like chest pains particularly during exercise indicated by ECG pseudo-infarction Q-waves, while 30% feel fatigued. Ventricular arrhythmias may cause palpitations. Although syncope and sudden cardiac death are common clinical outcomes in DCM patients, the do not always presents as the first symptom of DCM. Pulmonary and systemic thromboembolism are usually the first clinical manifestation of DCM [12]. The estimated mortality rate at the third year of the disease ranges between 12% and 20% [16]. There is no known cure for DCM with only cardiac transplantation showing short- and long-term favorable prognosis and survival rates [17].
Clinical Progression
Although clinical manifestation of DCM is known, determining the exact onset of these manifestations has remained challenging. It is common for DCM to manifest without clinical history or provoking factor [15]. More importantly, DCM has long sub-clinical phase that is asymptomatic, which decreases chances of detection. The subclinical phase, including intermediate phenotypes in the genetic transmission of DCM, also do not usually meet the standard diagnostic criteria making confirmatory diagnostic difficult [5]. (Figure 1).
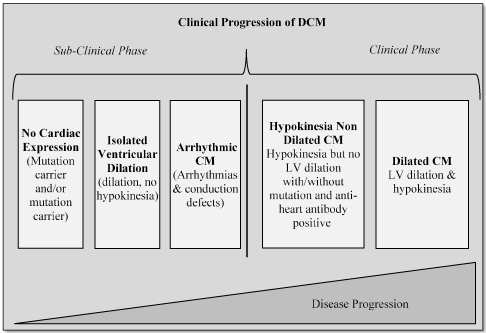
Figure 1. Clinical Progression of DCM
[Insert Figure 1 here]
In the subclinical phase, initially, DCM is asymptomatic, having no detectable LV abnormality nor arrhythmias. As DCM progresses, it begins to show symptoms of isolated ventricular dilation but no signs of hypokinesia, referring to a partial loss of muscle movement, which that do not fit the current definition of DCM and finally the presentation of arrhythmias and conduction abnormalities. In the clinical phase, hypokinesia develops in the absence of LV dilation. As the clinical phase progresses, patients develop both hypokinesia and LV dilation.
Prognosis
Status of Prognosis
Patients with DCM generally have an ominous prognosis. DCM is a frequent cause of heart failure and the primary indication for heart implantation [18]. In addition, DCM has a long sub-clinical phase presenting with or without symptoms and minor myocardial abnormalities, which increases the probability of missed early diagnosis and the institution of prompt therapeutic management [19]. In the subsequent clinical phase, the frequency of ventricular tachyarrhythmia in DCM patients has a negative correlation with survival rates, which requires chronic clinical management [20]. Patients with DCM have reduced survival rates of less than 50% at the tenth year of the disorder. However, with appropriate and adequate supportive care, patients can improve survival rates by between five and ten years [20] [21].
Despite the ominous prognosis, current clinical management methods have been demonstrated to improve prognosis. Medications such as angiotensin converting enzyme inhibitors (ACE-I), angiotensin II receptor blockers (ARB) and ß-blocker have been associated with improved survival rates of 80% and 65% at 5 and 10 years respectively. However, antiarrhythmic therapy indicated a statistically worse prognosis (p>0.05) compared to those without [21]. Heart implantation procedures such as implanted cardioverter defibrillator (ICD) produce the most favorable long-term prognosis for DCM patients. Heart transplantation is the only effective DCM therapy with a chronic protective role, improving survival rates by 87%, 71% and 55% at 10, 20 and 30 years respectively [20].
Predictors of Prognosis
Predictors of prognosis refer to factors or features of a disease used to assess or predict the progression of the disease or the efficacy of treatment. In DCM, the main predictors of prognosis are morphological features, clinical features and hemodynamic features [22].
Morphological Features
Morphological features refer to observable and measurable structural alterations in the left or both ventricles during the clinical progression of DCM. Ventricular dilation is the most distinguishing morphological feature of DCM and its severity is an independent predictor of prognosis [22]. A high degree of ventricular dilation suggests poorer prognosis, but the correlation is not linear. This is especially true in the case of a mildly dilated variant of DCM. In this variant, patients lack ventricular dilation but present with ventricular systolic dysfunction and a prognosis similar to patients with the dilated form of DCM [23] [24]. The LV ejection fraction (LVEF) is another independent predictor of prognosis. However, LVEF correlation with prognosis or survival is weaker in homogenous populations especially when LVEF ≤ 25% [25] [26]. Decreased ventricular mass-volume ratio, global wall motion abnormalities and near spherical LV cavity are other morphological features predicting poor prognosis [22].
Clinical Features
Clinical features refer to demonstrable or observable signs and symptoms of DCM at presentation or during treatment. The common clinical features predicting favorable prognosis include NYHA functional class > IV, young age and female gender. On the other hand, syncope, right-side heart failure and atrioventricular block or left bundle branch block on echocardiography predict poor prognosis. Cardiopulmonary exercises tests also provide prognostic information helpful in the evaluation of the extent of myocardial functional limitation [22].
Hemodynamic Features
Hemodynamics refer to alterations in the dynamics of blood flow observed in DCM patients. The common hemodynamic features or abnormalities that predict poor prognosis include pulmonary-capillary wedge pressure < 20mmHg, systemic hypotension, pulmonary hypertension and elevated cardiac pressure. However, the value of hemodynamic abnormalities to predict long-term prognosis before or after medical therapy remains unclear [27].
Risk Factors
Risk factors refer to individual characteristics, attributes or exposure to environmental factors such as toxins that increase the likelihood of developing or aggravating a disease condition when compared to the rest of the population [28]. Risk factors for DCM are continuous exposure to agents that interfere with the normal LV systolic function. These agents include genetic mutations, myocardial disorders and toxins [29]. Under genetic mutations, offspring of parents with non-ischemic heart failure and other myocardial abnormalities are at a greater risk of developing DCM because of the possibility of inheritance of the causative mutant genes [5]. Myocardial disorders such as myocardial ischemia is also a significant risk factor, and account for almost 50% of DCM. Other disorders such as coronary artery disease, hypertension and valvular disease that result in global systolic impairment also increases the likelihood of developing DCM [7]. Toxins such as excessive consumption of alcohol and chronic exposure to chemotherapeutic agents may predispose an individual to greater possibility of developing DCM. Finally, in pediatrics, inborn error of metabolism and malformation syndrome are also significant risk factors for developing DCM [14].
Pathophysiology
The hallmark of DCM is impaired LV systolic function caused by abnormal myocardial contractility [30]. The abnormal myocardium becomes unable to sustain normal systolic function and cardiac output. As a result, the LV and RV become overloaded with high cavitary blood volume, low ejection fraction and increased pressure causing them to dilate – stretch and to thin [4] [31]. Impaired LV systolic function may lead to RV-ventricular (bi-ventricular) systolic dysfunction. While LV dysfunction has been well established in DCM patients, recent studies suggest RV dysfunction is also prevalent in up to 65% of DCM patients [32]. Increased pressure leads to arterial ventricular valves to stretch and lose their synchrony causing blood to regurgitate into the atria. In turn, increased atrial pressure causes the atria to dilate resulting into increased pressure in the veins around the heart ultimately leading to heart failure, which is the final clinical endpoint of DCM [15].
Etiology
Etiology is a study of the cause of disease conditions useful for guiding therapeutic management [4]. The etiology of DCM is extremely heterogeneous. Half (50%) of the cases are idiopathic, caused mainly by inflammatory and immunological processes, while the other half results from a broad spectrum of underlying conditions, which includes peripartum disease, ischemic heart disease, myocarditis and hypertension [2]. Previously, DCM etiology was classified according to the causative agent: genetic/familial, cytotoxic agents, malnutrition, myocarditis/viral and autoimmune disorders. However, genetic abnormalities identified with familial DCM remains the most dominant etiology accounting for 20-48% of all DCM cases [15].
More recently, the ESC working group on myocardial and pericardial diseases reclassified the etiology of DCM under two main classes: genetic and non-genetic. Non-genetic etiologies include drugs/toxins, infection and peripartum. However, in some individuals, more than one etiologic agent may cause DCM. In such individuals, genetic agents interact with environmental (non-genetic) agents to cause DCM. Eliminating environmental agents is important to prevent the aggravation of DCM. The ESC working group on myocardial and pericardial diseases categorizes causes of DCM into genetic/familial, drugs/toxins, infection and peripartum [5] (Table 1).
Table 1. Causes and Agents of DCM
Group |
Cause |
Etiologic Agents |
Genetic/Familial |
Main Genes |
Titin, lamin A/C, myosin heavy chain, troponin, myosin-binding protein C, RNA-binding Motif-20, Myopalladin, Na+ channel alpha unit and phospholamban |
Neuromuscular Disorders |
Duchenne muscular dystrophy, Becker muscular dystrophy, myotonic dystrophy |
Syndromic Disease |
Mitochondrial disease, Tafazin |
Drugs/ Toxins |
Drugs |
Antineoplastic/psychiatric drugs |
Toxic Overload |
Ethanol, cocaine, amphetamines, ecstasy or iron overload |
Nutritional Deficiency |
Selenium, thiamine, zinc/copper and carnitine |
Electrolyte Disturbance |
Hypocalcemia, hypophosphatemia |
Endocrinology |
Hyper/hypo-thyroidism, Addison disease, phaeocromocytoma, acromegaly, diabetes mellitus |
Infection |
Auto-immune diseases (myocarditis) |
Causes frequent AV-block and ventricular arrhythmias |
Inflammatory DCM |
Caused by non-infectious myocarditis |
Peripartum |
Peripartum cardiomyopathy |
Related to during or after pregnancy |
[Insert Table 1 here]
Genetic Causes
DCM has a more pronounced heterogeneity in genetic etiology than any other cardiomyopathy phenotypes. Genetic etiologies consist of a variety of gene mutations in cytoskeleton, nucleoskeleton or mitochondrial proteins [7]. The primary pattern of genetic transmission is autosomal dominant. Inherited mutations in the sarcomere protein Titin (TTN) is the most frequent genetic cause of DCM, accounting for about 25% of familial DCM. Familial DCM refers to DCM inherited as a single mutated gene in a Mendelian pattern [33] [34]. Other common autosomal dominant genetic mutations are Lamin A/C, Myosin Heavy Chain, Troponin, Myosin-binding protein C, RNA-binding Motif-20, myopalladin, Na+ channel alpha unit, and Phospholamban [5] [35]. Although autosomal recessive mutations are a rare cause of DCM accounting for about 1-2% of familial DCM, increasing cases of X-linked recessive inheritance have been reported in tafazin gene in pediatric populations. Other X-linked recessive genetic causes are neuromuscular dystrophy and mitochondrial (syndromic) disease [6].
Non-Genetic Causes
Drugs/Toxins
The main non-genetic etiologic agents of DCM are drugs (also referred to as toxins), infection and peripartum DCM. Toxins, especially chronic or excessive alcohol consumption, or repeated exposure chemotherapeutic agents can induce DCM. Alcohol-induced cardiomyopathy causes the deterioration of LV systolic function and accounts for between 21% and 32% of DCM but reverses upon abstention [36]. Chronic exposure to some chemotherapeutic agents such as anthracylines can also affect LV function and induce DCM but upon withdrawal, either resolves by itself or persists in subclinical form [5].
Infection
Autoimmune viral infections such as myocarditis cause inflammations to induce DMC in genetically predisposed individuals. In some familial or non-familial patients, infection-negative myocarditis in the absence or presence of DCM phenotype is organ specific autoimmune disorder frequently found in genetically predisposed patients. These patients are asymptomatic but present with organ-specific anti-heart antibodies [37]. Anti-heart antibodies have been linked to mild LV abnormalities, which predicts DCM progression. In DCM caused by viral infection, if acute inflammation of myocardium stops and the cause resolves, and the disorder is reversible [38].
Peripartum
Peripartum cardiomyopathy (PPCM) is a rare myocardial condition affecting pregnant women or women who have just delivered. PPCM can induce or co-exist with DCM [39]. There are reported association of PPCM with ethnicity (Afro-Caribbean), age (older adults), multiple pregnancy and hypertension in the presence or absence of pre-eclampsia. PPCM as an etiology of DCM is complex, involving autoimmune disorders, viral infection, fetal microchimerism, stress-induced cytokines and toxicity due to abnormal production of prolactin [40].
Differential Diagnosis
The World Health Organization (WHO) and International Society and Federation of Cardiologists (ISFC), and more recently the ESC working group on myocardial and pericardial diseases developed the criteria for DCM diagnosis as follows:
- Fractional shortening >25% and/or LVEF < 45%;
- LV end-diastolic diameter > 117% corrected for age and body surface area in the exclusion of any known cause of myocardial disease.
- Diagnosis for familial DCM – the presence of more than one relative with DCM fitting the clinical criteria defined above or a relative of patient with DCM-related unexplained sudden death > 30 years [5] [12].
However, from a pathology standpoint, the current diagnostic criteria of DCM have important limitations. The criteria establish DCM as a non-specific phenotype resulting from genetic, acquired (environmental), both genetic and acquired or idiopathic etiologies [5].
Differential diagnosis is about distinguishing the cause of disease from others presenting with similar symptoms. It is clinically important in DCM to exclude other etiologies of cardiomyopathy with phenotypic overlap [2] [41]. It is also important to distinguish DCM from secondary causes of ventricular dilation and dysfunction caused by known cardiac or systemic processes such as ischemia, hypertension, valvular heart disease and myocarditis. Other secondary variants but less common include peripartum cardiomyopathy and those due to amyloidosis, sarcoidosis, and toxins such as doxorubicin [12]. In pediatric population, differential diagnosis is important to exclude metabolic cardiomyopathies, which occur at a higher frequency [42]. Figure 2 provides an illustration of diagnostic work-up (procedure) of DCM as proposed by the (ESC) working group on myocardial and pericardial diseases.
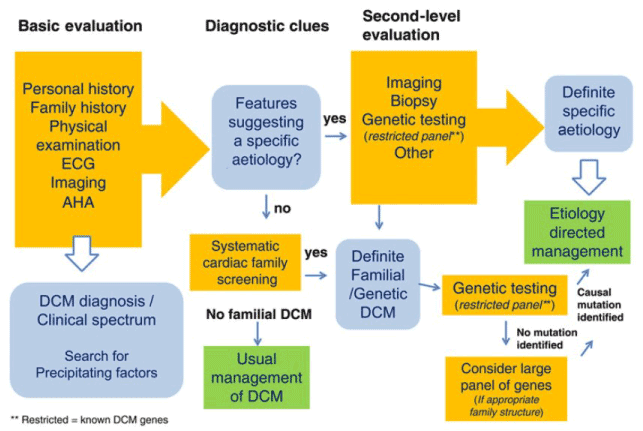
Figure 2. ESC Proposed Diagnostic Work-Up for DCM
[Insert Figure 2 here]
The DCM diagnostic process consists of clinical (first-line) and second-line diagnosis. Clinical diagnosis determines suspected DCM etiology based on patient/family history and physical signs and symptoms. The diagnostic clues from clinical evaluation guides rational selection of additional diagnostic tests in second line evaluation, which involves imaging, biopsy and genetic testing. The aim of second-line evaluation is to exclude other causes of myocardial disorder thereby providing a confirmatory diagnosis of DCM [5].
Patient/Family History
DCM is considered to have a genetic origin after the exclusion of secondary causes of ventricular dysfunction such as hypertension, myocarditis, valvular disease or exposure to toxins or environmental pathogens. Patient/family history provides valuable information for determining genetic origin of DCM. Patient/family history analysis begins with recording cardiac and extra cardiac patient history for those with suspected syndromic or metabolic DCM. The goal of analyzing patient history is to detect previous diagnostic clues such as myocardial disorders. The next step is to record detailed family history to identify family members suspected to have myocardial disorders or other diagnostic clues suggesting genetic etiology such as family history of heart failure, sudden cardiac death or heart implantation [43].
Analysis of family history involves constructing and analyzing a family pedigree of three or more generations. The primary objective is to determine the genetic transmission patterns: autosomal dominant, autosomal recessive, x-linked or matrilineal. Knowledge of genetic transmission pattern is significant to suggest the type of genetic mutation inherited and identification of relatives at risk of inheriting the genetic mutation. Diagnostic clues have also been suggested for various genetic transmission patterns [43] (Table 2).
Table 2. Genetic Transmission Patterns for DCM and Diagnostic Cues
Transmission Patterns |
Diagnostic Cues |
Autosomal Dominant |
- Male-to-male transmission
- Affected family members are in every generation
- Offspring have 50% risk of inheritance of mutated genes
- If father is affected, autosomal recessive/mitochondrial channels is excluded.
- If mother is affected, autosomal dominant inheritance is likely after exclusion of mitochondrial disease.
|
Autosomal Recessive |
- Suspected when both parents are carriers (not affected).
- Affects both male and females equally.
- Offspring have a 25% risk of inheritance
|
X-linked |
- Suspected when only/mostly males are affected.
- Daughters of affected fathers are carriers.
- No male-to-male transmission.
- More likely, if one/both parents have symptoms of skeletal muscles disorders.
|
Matrilineal |
- Mother transmits gene mutation to offspring
- Gene mutation usually in mitochondrial DNA.
- Affects both son/daughter.
- Suspected with abnormalities in different organs such as lactacidaemia, hypoacusia, encephalopathy
|
[Insert Table 2 here]
In addition to family history, pedigree analysis helps to identify other affected family members and refine diagnosis especially in families with more than one cardiomyopathy phenotypes. However, pedigree analysis does not identify de novo genetic mutation or myocardial disease unrecognized in the family tree [43] (Figure 3).
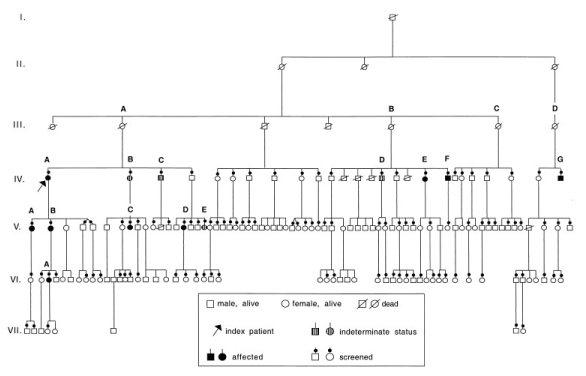
Figure 3. Pedigree Analysis of 7-Gerenartion of a White Family
[Insert Figure 3 here]
The pedigree analysis suggests an autosomal dominant pattern of genetic transmission (potentially excludes mitochondrial disease) with a mild form of familial DCM in which the onset of symptoms occurs in the fourth or later decades of life. Both the index patient and family members symptomatic of DCM have had favorable response to medical therapy. In addition to patient/family history, determining the age of first presentation is an important diagnostic clue for DCM. In infants, inborn errors of metabolism and dysmorphic syndromes are common causes compared to other age groups while coronary artery disease and transthyretin (TTR)-related amyloidosis is a is a common etiology in older adults [43]. Other non-cardiac autoimmune disorders such as diabetes mellitus (DM) can indicate underlying cause of inflammation about inflammatory DCM [5].
Physical Examination
Physical examination provides additional diagnostic clues about the underlying etiology of DCM. Physical examination determines physical signs and symptoms that could help distinguish DCM from other cardiomyopathy phenotypes. Routine physical examinations or specific inquiry is useful in the detection of cardiac involvement in cases of already diagnosed diseases such as Becker’s muscular dystrophy (BMD) and TTR amyloidosis where the presenting feature is cardiomyopathy [43]. Some common signs and symptoms checked for during physical examinations are learning difficulties and mental retardation, gait disturbance, visual impairment, skeletal muscle weaknesses and skin pigmentation (Table 3).
Table 3. Physical Examination – Physical Signs and Symptoms Suggesting DCM [40]
Signs and Symptoms |
Suspected DCM Etiology |
Learning difficulties/mental retardation |
Mitochondrial disease; Dystrophinopathies; Myotonic Dystrophy; TTN Mutation |
Sensorineural Deafness |
Mitochondrial Disease; Epicard mutation |
Visual Impairment |
Myotonic Dystrophy |
Gait Disturbance |
Dystrophinopathies |
Myotonia (delayed muscular relaxation) |
Myotonic Dystrophy |
Muscle weakness |
Dystrophinopathies; Myotonic Dystrophy;
Laminopathies; Desminopathy |
Skin pigmentation and scars |
Hemochromatosis |
[Insert Table 3 here]
Laboratory Tests
Laboratory tests provide additional diagnostic information about extra cardiac dysfunctions or disorders such as thyroid disease and diabetes mellitus that may contribute or exacerbate DCM. The test results help in the detection and assessment of secondary cardiac dysfunction, which are non-specific markers of DCM severity especially natriuretic peptides. The ESC recommends laboratory tests for all suspected cases of DCM including creatine phosphokinase (CK), renal function, proteinuria, red/white blood cells counts, serum iron, calcium phosphate and thyroid stimulating hormones. CK tests are diagnostically significant since raised CK is a diagnostic clue for X-linked dystrophin-related DCM. For mitochondrial-related DCM, laboratory test results indicating diagnostic suspicion are lactic acidosis, myoglobinuria and leukocytopenia [5]. Other important laboratory tests based on the suspected etiology include for serum antibodies, suspected infection (HIV, Chagas disease and influenza virus), thiamine for alcohol abuse or nutritional deficiency, and serum angiotensin converting enzyme to test for sarcoidosis [43].
Electrocardiogram
Electrocardiogram (ECG) is usually the initial cardiac screening test used for suggestive (non-confirmatory) diagnosis of DCM. The ESC recommends ECG to all first-degree relatives of the index patients. ECG test is non-specific, done to patients with definite or suspected cardiomyopathy. Abnormal ECG suggests phenotypic manifestation of a myocardial disorder. Its interpretation is usually in aggregation of findings from other tests such as echocardiography (echo) and cardiac magnetic resonance imaging (MRI) to confirm the underlying diagnosis of cardiomyopathy [1]. ECG abnormalities that raise suspicion for DCM are atrioventricular block, low P-wave amplitude, atrial standstill, low QRS voltage and right bundle branch bloc, or extremely low QRS amplitude [5].
Echocardiogram
Echocardiogram (echo) is the initial tool for imaging most phenotypes of myocardial disorders recommended for all first-line relatives of the index patient. However, echo cannot achieve differential diagnosis of DCM from LV dilation and dysfunction due to secondary etiologies. In two-dimensional (2D) echo, the degree of hypokinesia reveals segmental differences making distinction between DCM and ischemic cardiomyopathy difficult [15]. However, findings from echo are only useful when interpreted in the context of results from clinical and imaging results. In suspected DCM patients, three important features of echocardiogram tests are: (a) LV non-compaction for indication of genetic DCM of sarcomeric mutation; (b) postero-lateral dyskinesia/akinesia for indication for dystrophy-related DCM; and (c) dilatation and dyskinesia/akinesia for indication of myocarditis or sarcoidosis [43]. Although echocardiography has limited use in the confirmatory diagnosis of DCM, it provides several important diagnostic clues to increase clinical suspicion for DCM or for differential diagnosis of DCM (the exclusion of other potential cardiac and extra cardiac etiologies) [44,45] (Table 4).
Table 4. Key Echocardiographic Features in the Differential Diagnosis of DCM [45]
Diagnostic Clues |
Key Echocardiographic Features |
Idiopathic DCM |
Varying extent of dilation and dysfunction |
Ischemic Heart Disease |
Regional wall motion abnormalities/scar/aneurysm formation |
Hypertension |
LV Hypertrophy |
Severe Valvular Disease |
Valve abnormalities |
Infiltrative Disease (amyloid, sarcoidosis, hemochromatosis) |
Thickened myocardium, small pericardial effusion, focal aneurysm, abnormal myocardial texture |
Myocarditis |
None specific or small pericardial effusion |
[Insert Table 4 here]
Cardiac Catheterization
Cardiac catheterization is a medical procedure for diagnosing and treating cardiovascular disorders. The procedure involves inserting a catheter (thin flexible tube) in an artery or vein in the groin, neck or arm and then slowly it through the vein to the heart. In DCM, cardiac catheterization is used to exclude coronary artery disease (CAD) as the underlying cause of LV dilation and for the management of DCM. Cardiac catheterization also reveals high LV-end-diastolic pressure and pulmonary artery wedge pressure [15].
Cardiac MRI
The ability of cardiac MRI to assess cardiac morphology and functions enables the detection of specific forms of myocardial disease including differential diagnosis of ischemic and non-ischemic cardiomyopathies using late gadolinium-enhanced MRI. It is also able to characterize the presence and location of myocardial abnormalities based on their intrinsic magnetic properties and distribution of MRI contrast agent (gadolinium). The major diagnostic features examined by Cardiac MRI are LV enlargement/LV end-systolic volume, myocardial fat, iron storage, amyloid infiltration and myocardial fibrosis [2]. Various MRI sequences are used in DCM diagnosis. The common sequences include T1- and T2-weighted, T1-inversion recovery, contrast enhanced (late gadolinium), and spectroscopy [1] (Table 5).
Table 5. MRI Sequences Diagnostic Assessment of DCM [1]
MRI Sequence |
Description |
Features Targeted |
SSFP-Cine |
Regional/global biventricular dysfunction, ventricular mass/wall thickness |
Dilated LV/BV cavities; reduced EF<40%; ventricular wall thickness normal/reduced (<5.5mm) |
T2-W-STIR |
Increased myocardial free water |
Localized subendocardial hyper-intense signals distinguishing ischemic/non-ischemic disease |
IR-CE/LGE |
Myocardial fibrosis |
No enhancement, indistinguishable subendocardial from previous infarction, longitudinal mid-wall enhancement |
T1-Mapping |
Diffuse myocardial fibrosis |
T1 maps to characterize and quantify myocardial signal intensity |
MRS-Hydrogen |
Myocardial cellular triglyceride |
Insufficient research evidence |
MRS-Phosphorous |
Myocardial energetics |
Reduced PCr (~50%); ATP(~35%) & PCr/ATP(~25%) |
SSFP: Steady-State Fast Precession; T2-W: T2-weighted; STIR: Short-T1 Inversion Recovery; IR-CE: Inversion Recovery Contrast-Enhanced; LGE: Late Gadolinium; MRS: Magnetic Resonance Spectroscopy; PCr: Phosphocreatine; ATP: Adenosine-5-triphosphate
[Insert Table 5 here]
Nuclear Imaging
The contribution of nuclear imaging to the diagnosis of cardiomyopathies is limited except in the diagnosis and prognosis of sarcoidosis and TTR-associated amyloidosis [43]. Previously, thallium-201 and gallium-67 radionuclide scintigraphy provided diagnostic and prognostic assessment of sarcoidosis [46]. Accumulated gallium-67 is an indication of active inflammation associated with sarcoidosis or acute myocarditis. Positron emission tomography (PET) measurement of the uptake of 18F-?uorodeoxyglucose (18F-FDG) has high sensitivity for sarcoidosis but also seen in idiopathic DCM and healthy tissues resulting into many false positives. However, PET 18F-FDG uptake in myocardial tissues is suggestive of sarcoidosis. Nuclear imaging using 99m Tc-3,3-diphosphono-1,2-propanodi-carboxylic acid scintigraphy can detect TTR amyloid mutation but not in sarcomeric-induced hypertrophic cardiomyopathy [47].
Meta-Analysis of Differential Diagnosis
Search Criteria
The current meta-analysis combines research findings on diagnosis of DCM to advance knowledge of the common diagnostic methods and key diagnostic features of DCM. The search for primary references and reviews was carried on three electronic databases: PubMed, EMBASE and Cochrane as well as in Google Scholar. A combination of search terms used were diagnosis OR echocardiography OR cardiac magnetic resonance AND dilated cardiomyopathy (DCM). There was no restriction on publication date or language. Studies were included irrespective of the age of the patients – both adults and pediatric patients were eligible for inclusion.
Study Selection
Studies were selected if they (a) utilized at least on diagnostic methods – both imaging and non-imaging methods; (b) assessed patients suspected with DCM; (c) provided information on diagnostic or prognostic outcomes. Additional studies ewer retrieved from bibliographies of included studies and from review studies and subjected to the inclusion criteria. Studies available only in abstract form (without a published manuscript) or data was not readily extractable were excluded. Data was extracted from each of the included studies and recorded in Microsoft Excel Worksheet. The extracted data included name of first author and year of publication, number of patients recruited, diagnostic method used, diagnostic feature targeted and the summary of main findings (Table 6).
Table 6. Summary of Data from the Included Studies on DCM Diagnosis/Prognosis
1 st Author (year) |
Sample |
Diagnostic Method |
Diagnostic Feature |
Summary of the Main Findings |
Andreini et al. (2007) [48] |
61 |
Multi-Detector CTA (16-Slice) |
Degree of stenosis |
It is a feasible and safe method for the identification of idiopathic vs. ischemic DCM based on the degree of stenosis. |
Assomul et al. (2006) [49] |
101 |
CMR (Siemens Sonata 1.5T) |
Midwall myocardial fibrosis or Midwall LGE |
CMR-Midwall fibrosis predicts SDC, VT, cardiac hospitalization. Helpful for risk stratification and selection for those in need of therapy. |
Dec et al. (1990) [50] |
82 |
Indium-111-labeled antimyosin antibodies |
Myocyte necrosis (component of myocarditis) |
Useful for the evaluation of patients with dilated or non-dilated cardiomyopathy suspected with myocarditis. |
Halliday et al. (2017) [51] |
LGE-CMR (Siemens Sonata 1.5T) |
Midwall LGE and SCD |
Midwall LGE identifies DCM and LVEF≥50% at risk of SCD and those to benefit from ICD. |
Kotani et al. (2016) [46] |
1 |
SPECT/CT |
Atrial wall |
Useful for detection of acute myocarditis or sarcoidosis. |
Machii et al. (2014) [52] |
72 |
LGE-CMR (Philips Achieva 1.5T) |
LGE and SCD |
Detects none, localized and extensive LGE. Extensive LGE has no functional recovery and lowest event-free survival rate. Patients with no/localized LGE have reverse remodeling after treatment. |
Marra et al. (2014) [53] |
137 |
LGE-CMR (Siemens Sonata 1.5T). |
Myocardial fibrosis |
Presence of LV-LGE is an independent predictor of malignant arrhythmias. Amount and distribution have no prognostic value. |
McCrohon et al. (2003) [54] |
90 |
LGE-CMR |
Midwall LGE |
Differentiates DCM from CAD-related LV dysfunction. |
O'connell et al. (1984) [47] |
68 |
Nuclear (radio-isotopic) imaging |
Gallium-67 |
Feasible and safe test to identify patients which myocarditis on biopsy and minimizes the need for frequent biopsies |
Pinamonti et al. (1993) [55] |
79 |
Doppler echocardiography |
Restrictive LV filling, E-deceleration |
Restrictive LV filling pattern is frequent in DCM associated with worse prognosis, indicator for high mortality risk and the need for heart transplantation. |
Puntmann et al. (2013) [56] |
82 |
T1 native and post-contrast (Philips Achieva 3T) |
Comparing normal vs diffuse myocardial fibrosis |
T1 was longer in DCM patients while post-contrast was shorter then in normal myocardium. T1 native distinguished normal from diseased myocardium. |
Rihal et al. (1994) [30] |
102 |
2-Dimension and Doppler Echocardiography |
LV filling pattern, deceleration time (DT), ejection fraction (EJ) |
Diastolic dysfunction correlated with congestive symptoms while systolic dysfunction was an indicator of survival. EJ and DT identified divergent chronic prognosis. |
Sato et al. (2001) [57] |
60 |
Immunoassay kit (Roche Diagnostics |
Serum concentration of TTN |
TNT is a sensitive marker for myocardial injury. It detects myocyte industry and an indicator for poor prognosis |
Sharp et al. (1994) [58] |
70 |
Dobutamine stress echocardiography |
Ventricular wall motion |
Allows diagnostic of CAD as cause for LV dysfunction other than CAD. |
CMR: Cardiac Magnetic Resonance; SDC: Sudden Cardiac Death; VT: Ventricular Tachycardia; LGE: Late Gadolinium Enhancement: ICD: Implantable Cardioverter Defibrillator; SPECT/CT: Single Photon Emission Computed Tomography/Computed Tomography (SPECT/CT)
[Insert Table 6 here]
Study Characteristics
After screening all the potential studies against the inclusion criteria, fourteen (14) studies investigating DCM diagnosis were included in the present meta-analysis [30,46-58]. The studies were published between 1994 and 2017. In total, the studies recruited 1,314 patients suspected with DCM. Different diagnostic methods were used to reveal important diagnostic clues or to exclude other potential causes of dilated myocardium. The most common diagnostic method for DCM was non-invasive cardiac imaging. Cardiac MRI was the most common imaging technique studied by 43% of the included studies [49,51,52,53,54,56]. The most frequently used imaging modalities were LGE-MRI and T1-weighted or post-contrast. Cardiac MRI was useful for comparing normal versus diffuse myocardial fibrosis or for characterizing midwall myocardial fibrosis or midwall LGE. The second frequently used imaging technique was Doppler echocardiography studied by 21% of the studies [30,55,58] used for characterizing myocardial hemodynamics – LV filling pattern, deceleration time (DT), ejection fraction (EJ) and ventricular wall motion. Cardiac CT angiography (SPECT or Multi-Detector CTA) was an uncommon imaging technique investigated by 14% of the studies. It was useful for assessing the extent of stenosis [46,48]. Finally, nuclear imaging and laboratory tests were less common studied by 14% and 7% of the studies respectively.
Study Outcomes
Cardiac LGE-MRI was the most used cardiac imaging modality for the diagnosis and prognostication of DCM in suspected patients [49,51,52,53,54]. It was valuable in assessing both ventricular abnormalities and prognostication. Cardiac LGE-MRI enables the detection of midwall fibrosis (defined as none, localized or extensive LGE) as well as the exclusion of CAD-related LV dysfunction from DCM [49,51,52,54]. It is valuable in providing prognostic information and choice of the most appropriate treatment. Extensive midwall LGE indicates a significantly reduced likelihood of ventricular functional recovery and the lowest event free survival while patients with none or localized midwall LGE have higher probability of reverse modeling after treatment [52]. The presence of LV-LGE or significantly depressed LV function (LVEF ≥ 50%) predicts an elevated risk of sudden cardiac death and an indication for device therapy (ICD) [51]. LV-LGE also suggests an elevated risk of malignant ventricular arrhythmias, cardiac hospitalization and risk stratification for patients in need or chronic therapies [49,53].
Echocardiography was the second most common imaging technique for assessing myocardial alterations in patients suspected with DCM. However, unlike cardiac MRI that was largely used to assess morphological myocardial alterations, Doppler echocardiography was used to assess functional alterations [30,55,58]. It enabled the detection of restrictive LV filling patterns, depressed ejection fraction and E-deceleration [30,55]. Dobutamine stress echocardiography assessed ventricular wall motion and enabled differentiation from LV dysfunction due to DCM from that due to CAD. In addition to DCM diagnosis, Doppler echocardiography predicts a worse prognosis based on restrictive ventricular filling, an indicator for high mortality rate and a frequent indication for cardiac transplantation [55] while systolic dysfunction is an indicator for survival [30]. Cardiac CT angiography is less common in the diagnosis of DCM. However, Multi-Detector CTA provides valuable information on the extent of stenosis and a promising method to distinguish idiopathic from ischemic DCM [48] and SPECT provides information on alterations in arterial wall and enables differentiation of acute cardiac myocarditis or cardiac sarcoidosis from DCM [46].
The use of other tests such as nuclear imaging is very uncommon in the diagnosis of DCM. However, nuclear imaging using Gallium-67is feasible and safe for testing myocarditis, which minimizes the need for frequent biopsies [47]. In addition, nuclear imaging using Indium-111-labeled antimyosin antibodies indicates myocyte necrosis, useful for evaluation of dilated and non-dilated cardiomyopathy in patients suspected with myocarditis [50]. Finally, laboratory tests provide information on important biomarkers such as TNT, which have high sensitivity in detecting myocardial damage [57].
Discussion
The present findings reveal the mainstay of diagnosis of DCM is cardiac imaging, which enables the characterization of morphological and/or functional ventricular abnormalities. Cardiac LGE-MRI and Doppler echocardiography were clinically effective in assessing both morphological and functional myocardial or ventricular alterations suggestive of DCM-related myocardial damage as well as in differentiation LV dysfunction due to DCM and that due to other causes such as coronary artery disease and myocarditis. Cardiac MRI and echocardiography also provided clinically valuable prognostic markers for identifying patients at higher likelihood of achieving normal LV function after therapy, and for stratifying patients at risk of malignant ventricular arrhythmias and sudden cardiac death. They were also clinically valuable for selecting patients for chronic therapy such as device therapy and cardiac transplantation. Other tests providing supplementary diagnostic clues were nuclear imaging, cardiac CT angiography and laboratory tests for cardiac biomarkers.
The present findings are consistent with WHO/ISFC and ESC recommendations of diagnosis of DCM. The three professional cardiology associations recommend cardiac imaging of alterations in the function and structure of the myocardium are the cornerstone of DCM diagnosis [5,12]. In the recommendations, diagnosis of DCM involves assessment of myocardial dysfunction defined as fractional shortening > 25% or LVEF < 45%, and/or LV end-diastolic diameter > 117% using cardiac MRI [5]. Previous studies have also demonstrated the value of cardiac LGE-MRI in the diagnosis of DCM. It enables the visualization of DCM-related myocardial dysfunction including the differentiation of ischemic and non-ischemic DCM, and the detection of the location and extent of myocardial abnormalities. Cardiac LGE-MRI provides additional diagnostic clues by detecting LV enlargement, myocardial fat, iron storage, amyloid infiltration and myocardial fibrosis [2].
On the other hand, although the present findings suggest the value of echocardiography in the diagnosis of DCM, its use is limited by its inability to differentiate DCM-related LV dysfunction and LV dysfunction due to secondary etiologies [15]. In addition, the extent of hypokinesia shows segmental differences in 2D-echocardiography limiting the ability to differentiate non-ischemic DCM from ischemic cardiomyopathy [12]. However, Doppler echocardiography remains valuable in assessing restrictive ventricular filling associated with DCM. In addition to cardiac MRI and echocardiography, nuclear imaging may be used in the diagnosis of DCM. However, it has limited use except in the detection of cardiac sarcoidosis and TTR-related cardiac amyloidosis [43]. Besides imaging laboratory tests for cardiac biomarkers associated with LV dysfunction and the identification of cardiac dysfunction from extra cardia disorders such as thyroid disease and diabetes mellitus [5]. ECG abnormalities also suggest myocardial abnormalities and family history provides clues on genetic etiologies of DCM [5].
In summary, the basis of diagnosis of DCM is the assessment of ventricular dilation and depressed myocardial function and the exclusion of other potential etiologies such as hypertension, valvular, congenital or ischemic heart disease. The cornerstone of diagnosis is cardiac LGE-MRI that is able to detect the location and extent of myocardial damage and differentiate ischemic from non-ischemic DCM. Doppler echocardiography is useful to detect restrictive ventricular filling patterns while laboratory tests and family screening is essential for providing clues for the involvement of extra cardiac causes and the genetic basis of DCM respectively.
Clinical Management
According to the 2016 AHA recommendations and diagnosis of dilated cardiomyopathies [8], clinical management of DCM lacks specific etiology-based therapy. Instead, treatment draws upon the general management concepts of LV dysfunction and heart failure therapy. Current therapy includes general (environmental) measures, conventional pharmacotherapy, mechanical devices, and genetic counselling [11].
General Measures
General measures target to reduce the exposure to environmental factors that may aggravate DCM phenotype by burdening the susceptible myocardium. General measures frequently prescribed to DCM patients include patient education, restriction on fluids and salt, treating hypertension, stop or limit alcohol consumption, management of body weight, and moderate aerobic exercise [11].
Pharmacotherapy
Pharmacotherapy is not specific to etiopathogenesis but shows favorable prognosis and a reduction in mortality [17,18]. Pharmacotherapy includes several drugs adopting concepts and standards used for the management of heart failure as discussed in the subsequent questions [59].
Angiotensin Converting Enzyme Inhibitors
Several randomized controlled trials (RCTs) including CONSENSUS, SOLVD and SAVE have demonstrated the prognostic value of Angiotensin Converting Enzyme Inhibitors (ACEI) in reducing the progression, hospitalization and mortality of heart failure. Prescription begins at low doses, slowly increased up to doses demonstrated in RCTs as efficacious. For instance, the maximum daily dose for Captopril is 50 mg thrice, Enalapril 20 mg twice and Lisinopril 40 mg once [11].
Angiotensin II Receptor Blockers
For patients intolerant to ACE-Inhibitors, Angiotensin II Receptor Blockers (ARB) are a promising alternative. Several RCTs, ELITE, Val-HeFT and OPTIMAAL have shown ARB are safe and effective but its addition to ACEI does not provide any additional clinical benefit [1]. Some ARBs such as first-generation calcium channel blockers and endothelin antagonists are not recommended in standard guidelines for heart failure [60].
Beta-Adrenergic Blockers
Beta-Adrenergic Blockers have shown efficacy for management of supraventricular and ventricular arrhythmia. RCTs have reported Metoprolol and Bisoprolol reduced in all-cause mortality by 34% and Carvedilol, which has alpha-blocking properties, by 35% for severe heart failure. Beta-blockers begin with lower doses and titrated gradually to the target dose 25 mg or 50 mg twice a day depending on patient weight [11,18]. However, long-term effect of beta-blocker does not reduce the mortality due to sudden cardiac death [14].
Heart Transplantation
Heart transplantation is the only long-term therapy improving survival rates of DCM patients [17]. ICD and bi-ventricular pacemakers are long-term therapies for preventing sudden cardiac death in selected genetic and acquired (non-genetic) DCM with LV dysfunction. For selected DCM patients with pro-longed QRS duration and LV dysynchrony, a combination of ICD and cardiac resynchronization therapies are indicated [57]. ICD has beneficial outcomes for DCM patients with ischemic-induced systolic dysfunction but no significant reduction in mortality in non-ischemic DCM [61,62].
Meta-Analysis of ICD Therapy in Prevention of DCM-Related Deaths
Chronic clinical management of DCM has focused on device therapy, with implantable cardioverter defibrillator (ICD) being a frequent indication for DCM patients with significantly depressed LV function and symptomatic heart failure. The primary indication of ICD therapy is the prevention of sudden cardiac death in idiopathic DCM and selected secondary DCM. However, convincing evidence lacks on the protective value of ICD against cardiac death in DCM patients with some studies suggesting no significant reduction in mortality [61,62]. The present meta-analysis combines research outcomes on the clinical efficiency of ICD compared to medical therapy in the prevention of cardiac death in DCM patients.
Search Strategy/Inclusion
Prospective randomized trials investigating the use of ICD therapy on DCM patients were searched using a two-level search strategy: online and manual search. In the first level, online databases PubMed, EMBASE and MEDLINE were searched using a combination of keywords. A combination of broad-based key words were used, which included implantable cardioverter defibrillator, randomized controlled trials, clinical trials, non-ischemic dilated cardiomyopathy, and sudden cardiac death. Studies were considered for inclusion if they met the following inclusion criteria: (a) was a prospective randomized controlled trials; (b) recruited patients diagnosed with non-ischemic DCM; (c) patients were randomly assigned to ICD therapy; and (d) the main outcomes included cardiac death, all-cause mortality and arrhythmic death. In the second level, relevant studies were identified through a manual search of secondary sources including bibliographies of initially identified articles and review articles. There was no restriction on publication year or language. All references were downloaded for consolidation and removal of duplicates for further screening.
Data Assessment/Abstraction
All potential studies were assessed for quality using a modified version of the Oxford quality scoring system (Jadad scale) [14] was used to assess the quality of the included studies [63]. Scoring involved 11 questions with a total score of 13 points (two point for the first two questions). The 11 questions were. (i) Was the study described as randomized? (ii) Was there concealment of randomisation? (iii) Was there a description of withdrawals and dropouts? (iv) Were study objective defined? (v) Were outcomes measured and define clearly? (vi) Was there a clear description of inclusion and exclusion criteria? (vii) Was the patient sample justified? (viii) Was there a clear description of interventions used? (ix) Was there a control group? (x) Were methods assessing adverse effects clearly described? (xi) Were statistical methods clearly described and justified? After quality assessment, each study was screened for inclusion criteria and subsequently data collated from all the included studies. The extracted data was summarized in a Microsoft Excel Spreadsheet. The extracted data included name of first author, number of patients recruited, mean age, percentage of male patients, mean LVEF at presentation, mortality rate, NYHA functional class III/IV percentage and mean duration of follow up (Table 7).
Table 7. Summary of Data on ICD Therapy for DCM from the Included Studies
1st Author Name [Ref] |
Patient Sample Size |
Mean Age (yrs.) |
% Male |
LVEF (%) |
Mortality Rate (1yr) |
NYHA III/IV (%) |
Mean F/Up (Months) |
Defibrillators [64] |
193 |
65 |
79 |
32 |
18 |
10 |
18 |
Kuck et al. [65] |
36 |
58 |
80 |
46 |
15 |
18 |
57 |
Connolly et al. [66] |
63 |
64 |
85 |
34 |
10 |
11 |
36 |
Bänsch et al [67] |
104 |
52 |
80 |
24 |
4 |
35 |
66 |
Strickberger et al [68] |
103 |
59 |
70 |
23 |
10 |
20 |
24 |
Kadish et al. [69] |
458 |
58 |
71 |
21 |
6 |
21 |
26 |
Bardy et al. [70] |
792 |
60 |
77 |
25 |
7 |
30 |
46 |
Bristow et al. [71] |
397 |
67 |
68 |
22 |
19 |
100 |
16 |
[Insert Table 7 here]
Study Characteristics
Eight (8) prospective randomized controlled trials (RCTs) meeting the inclusion criteria were included in this meta-analysis [64-71]. In total, the eight studies recruited 2,146 patients diagnosed with non-ischemic dilated cardiomyopathy. Patient population was largely male (76%) with a mean age of 60 years range 52 to 67 years. The patients were followed up in a mean period of 36 months, range 18 to 56 months for clinical outcomes of ICD therapy. The eight studies investigated ICD therapy either alone or use concomitant with cardiac resynchronization therapy (CRT) compared with conventional HF therapy (ACE-inhibitors/Angiotensin-Receptor Blocker (ARB) and beta-blocker).
Study outcomes
Three studies [64-66] investigated ICD as a secondary therapy after medical therapy failed to achieve the intended clinical outcomes (reversed LV function or relived HF symptoms). Individually, the three studies indicate ICD had better protective outcomes compared to medical therapy against cardiac death but the difference was not statistically significant. The remaining five studies [67-71] investigating ICD as the primary therapy also reveal ICD has a better protective effect against cardiac death but individually the difference was not significant. The present findings reveal ICD therapy either alone or a dual therapy with CRT conveys a protective effect against cardiac death on DCM patients presenting with significantly depressed LV function (LVEF < 25%) and symptoms of heart failure. The mean one-year mortality rate was estimated at 11%. When the studies are pooled together, ICD therapy has a statistically superior protective effect over medical therapy against cardiac death and all-cause mortality in non-ischemic DCM patients.
Discussion
Clinical management of DCM is limited by the lack of specific etiology-based therapy. Current short-term therapies rely on conventional medication for heart failure, while chronic therapy relies on device therapy (ICD and/or cardiac resynchronization therapy) and heart transplantation [8]. The basis of ICD therapy is its clinical efficacy in both primary and secondary prevention of sudden cardiac death in patients diagnosed with myocardial infarction and significantly depressed LV function. However, the value of ICD on protecting against sudden cardiac death in non-ischemic DCM patients has not been well established, with individual studies suggesting an insignificant value. However, the present meta-analysis finds ICD reduces the risk of cardiac death with one-year mortality at 11%. It has a superior long-term primary and secondary protective effect against cardia death when compared to medical therapy in selected sub-groups of DCM patients, principally, those with significantly depressed LVEF < 28% and NYHA Functional Class III or IV.
The value of ICD therapy on improving LV dysfunction secondary to ischemic cardiomyopathy has been well established but its value on improving DCM-related LV dysfunction or as a prophylactic against cardiac death is an ongoing research area [8]. Although the efficacy of ICD therapy on mortality reduction has received research support, its use may not suggest a greater benefit for DCM patients with end-stage heart failure [61,62]. Indication for ICD therapy requires careful consideration for individual preferences of DCM patients and their perception of quality of life. In addition, while ICD reduces the risk of sudden cardiac death, there is also no research evidence of ICD preventing the progression of heart failure [8]. For DCM patients presenting with both severe LV dysfunction and specific ECG abnormalities such as prolonged QRS duration, a dual therapy of ICD and CRT may produce a greater prophylactic effect against cardiac death [62]. In summary, ICD therapy is more effective compared to medication in both primary and secondary prevention of cardiac death in sub group of DCM patients with significantly depressed LV function and symptomatic heart failure. However, ICD therapy does not prevent the progression of heart failure or has limited benefits for DCM patients with end stage heart failure.
Conclusion
Dilated cardiomyopathy (DCM) is a progressive, and usually, irreversible heterogenous myocardial disorders characterized by left ventricular or bi-ventricular dilation, impaired systolic function, myocyte degeneration and interstitial fibrosis not explained by coronary artery disease (CAD) or abnormal hemodynamic overload. It is the most prevalent cardiomyopathy phenotype, with higher reported cases in adults and males. Clinically, DCM progresses from asymptomatic sub-clinical phase to symptomatic clinical phase. The initial clinical manifestations are pulmonary and systemic thromboembolism. Other more frequent clinical signs and symptoms are significantly depressed LV function, New York Heart Association (NYHA) functional class III to IV, exertional angina, ventricular arrhythmias and syncope. Predictors of poor prognosis include ventricular dilation (LVEF ≤ 25%), right-sided heart failure and restrictive hemodynamics. Risk factors for developing DCM is the continuous exposure to agents interfering with LV function such as genetic mutations, myocardial disorders and toxins. DCM has heterogeneous etiologies broadly categorized into idiopathic etiologies occurring secondary to inflammatory and immunological processes, and secondary etiologies occurring in the setting of a broad spectrum of cardiac and extra cardiac conditions such as peripartum disease, ischemic heart disease, myocarditis and hypertension. Diagnosis of DCM relies on the detection of structural and functional myocardial abnormalities using cardiac LGE-MRI and Doppler echocardiography complemented with family screening to detect genetic basis of DCM and laboratory test to detect extra cardiac causes. Finally, clinical management does not have a specific etiology-based therapy. However, pharmacotherapy has shown favorable prognosis in the short-term while heart transplantation and device therapy (ICD and bi-ventricular pacemakers and/or cardiac resynchronization) have shown favorable long-term prognosis and survival rates.
References
- Bowles NE (2002). The molecular biology of dilated cardiomyopathy. Eur Heart J Suppl, 4(suppl_I), I2-I7.
- Francone M (2014). Role of cardiac magnetic resonance in the evaluation of dilated cardiomyopathy: diagnostic contribution and prognostic significance. ISRN Radiology, 2014:365404. [Crossref]
- Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, et al (1996) Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation, 93, 841-842. [Crossref]
- Gulati A, Ismail TF, Jabbour A, Alpendurada F, Guha K, et al (2013). The prevalence and prognostic significance of right ventricular systolic dysfunction in non-ischemic dilated cardiomyopathy. Circulation, 113, 1-37.
- Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, et al. (2016). Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J, 37: 1850-1858. [Crossref]
- Merlo M, Gentile P, Naso P, Sinagra G (2017). The natural history of dilated cardiomyopathy: how has it changed? J Cardiovasc Med (Hagerstown)., 18, e161-e165. [Crossref]
- McNally EM, Golbus JR, Puckelwartz MJ (2013) Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest 123: 19-26. [Crossref]
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A, et al. (2016). Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation, 134(23), e579-e646. [Crossref]
- Honjo K (2004). Social epidemiology: Definition, history, and research examples. Environmental Health and Preventive Medicine, 9: 193-199. [Crossref]
- World Health Organization. (2017). Epidemiology fact sheet. Available at http://www.who.int/topics/epidemiology/en/
- Rakar S, Sinagra G, Di Lenarda A, Poletti A, Bussani R, et al. (1997) Epidemiology of dilated cardiomyopathy. A prospective post-mortem study of 5252 necropsies. The Heart Muscle Disease Study Group. Eur Heart J 18: 117-123. [crossref]
- Taylor MR, Carniel E, Mestroni L (2006) Cardiomyopathy, familial dilated. Orphanet J Rare Dis 1: 27. [crossref]
- Codd MB, Sugrue DD, Gersh BJ, Melton LJ 3rd. (1989). Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975-1984. Circulation, 80: 564-572. [Crossref]
- Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, et al. (2006) Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 296: 1867-1876. [crossref]
- Sisakian H (2014) Cardiomyopathies: Evolution of pathogenesis concepts and potential for new therapies. World J Cardiol 6: 478-494. [crossref]
- Goldberger JJ, Suba�ius H, Patel T, Cunnane R, Kadish AH (2014). Sudden cardiac death risk stratification in patients with nonischemic dilated cardiomyopathy. Journal of the American College of Cardiology, 63: 1879-1889. [Crossref]
- Tardiff JC, Carrier L, Bers DM, Poggesi C, Ferrantini C (2015). Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc Res, 105: 457-470. [Crossref]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, et al. (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 113: 1807-1816. [Crossref]
- Arbustini E1, Narula N2, Tavazzi L3, Serio A1, Grasso M1, et al. (2014) The MOGE(S) classification of cardiomyopathy for clinicians. J Am Coll Cardiol 64: 304-318. [Crossref]
- Merlo M, Pivetta A, Pinamonti B, Stolfo D, Zecchin M, et al. (2014). Long-term prognostic impact of therapeutic strategies in patients with idiopathic dilated cardiomyopathy: Changing mortality over the last 30 years. Eur J Heart Fail., 16: 317-324. [Crossref]
- Matsumura Y, Takata J, Kitaoka H, Kubo T, Baba Y, et al. (2006). Long-term prognosis of dilated cardiomyopathy revisited. Circ J, 70: 376-383. [Crossref]
- Dec GW, Fuster V (1994) Idiopathic dilated cardiomyopathy. N Engl J Med 331: 1564-1575. [crossref]
- Gavazzi A, De Maria R, Renosto G, Moro A, Borgia M (1993). The spectrum of left ventricular size in dilated cardiomyopathy: clinical correlates and prognostic implications. Am Heart J., 125: 410-422. [Crossref]
- Keren A, Gottlieb S, Tzivoni D, Stern S, Yarom R, et al. (1990). Mildly dilated congestive cardiomyopathy. Use of prospective diagnostic criteria and description of the clinical course without heart transplantation. Circulation, 81: 506-517. [Crossref]
- Griffin BP, Shah PK, Ferguson J, Rubin SA (1991). Incremental prognostic value of exercise hemodynamic variables in chronic congestive heart failure secondary to coronary artery disease or to dilated cardiomyopathy. Am J Cardiol., 67: 848-853.
- Saxon LA, Stevenson WG, Middlekauff HR, Fonarow G, Woo M, et al. (1993). Predicting death from progressive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol, 72: 62-65.
- Keogh AM, Baron DW, Hickie JB (1990). Prognostic guides in patients with idiopathic or ischemic dilated cardiomyopathy assessed for cardiac transplantation. Am J Cardiol, 65: 903-908.
- Unverferth DV, Magorien RD, Moeschberger ML, Baker PB, Fetters JK (1984). Factors influencing the one-year mortality of dilated cardiomyopathy. Am J Cardiol, 54: 147-152. [Crossref]
- Haas J, Frese KS, Peil B, Kloos W, Keller A, et al. (2014). Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J , 36: 1123-1135. [Crossref]
- Rihal CS, Nishimura RA, Hatle LK, Bailey KR, Tajik AJ (1994). Systolic and diastolic dysfunction in patients with clinical diagnosis of dilated cardiomyopathy. Relation to symptoms and prognosis. Circulation, 90: 2772-2779.
- Belloni E, De Cobelli F, Esposito A, Mellone R, Perseghin G, et al. (2008) MRI of cardiomyopathy. AJR Am J Roentgenol 191: 1702-1710. [crossref]
- La Vecchia L, Zanolla L, Varotto L, Bonanno C, Spadaro GL (2001). Reduced right ventricular ejection fraction as a marker for idiopathic dilated cardiomyopathy compared with ischemic left ventricular dysfunction. Am Heart J, 142: 181-189. [Crossref]
- Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC (2015). Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science, 349: 982-986. [Crossref]
- Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, et al. (2014). The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med., 16: 601-608.
- Hanley A, Walsh KA, Joyce C, McLellan MA, Clauss S, et al. (2016). Mutation of a common amino acid in NKX2. 5 results in dilated cardiomyopathy in two large families. BMC Medical Genetics, 17: 83. [Crossref]
- George A, Figueredo VM (2011) Alcoholic cardiomyopathy: a review. J Card Fail 17: 844-849. [crossref]
- Caforio AL, Mahon NG, Baig MK, Tona F, Murphy RT, et al. (2007). Prospective assessment in dilated cardiomyopathy: cardiac autoantibodies predict disease development in asymptomatic relatives. Circulation 115,115:76-83. [Crossref]
- Mahon NG, Murphy RT, MacRae CA, Caforio AL, Elliott PM, et al. (2005). Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Ann Intern Med 143, 108-111. [Crossref]
- van Spaendonck-Zwarts KY, van Tintelen JP, van Veldhuisen DJ, van der Werf R, Jongbloed JD, et al. (2010). Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation, 121: 2169-2175. [Crossref]
- *Hil?ker-Kleiner D, Haghikia A, Nonhoff J, Bauersachs, J (2015). Peripartum cardiomyopathy: current management and future perspectives. Eur Heart J, 36: 1090–1097. [Crossref]
- Lima JA, Desai MY (2004). Cardiovascular magnetic resonance imaging: current and emerging applications. J Am Coll Cardiol., 44: 1164-1171. [Crossref]
- Mestroni L, Rocco C, Gregori D, Sinagra G, Di Lenarda A (1999). Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. J Am Coll Cardiol., 34, 181-190. [Crossref]
- Rapezzi C, Arbustini E, Caforio AL, Charron P, Gimeno-Blanes J, et al. (2012). Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J., 34: 1448-1458. [Crossref]
- Crispell KA, Wray A, Ni H, Nauman DJ, Hershberger RE (1999). Clinical profiles of four large pedigrees with familial dilated cardiomyopathy: Preliminary recommendations for clinical practice. J Am Coll Cardiol., 34: 837-847. [Crossref]
- Thomas DE, Wheeler R, Yousef ZR, Masani ND (2009). The role of echocardiography in guiding management in dilated cardiomyopathy. Eur J Echocardiogr., 10: iii15-iii21. [Crossref]
- Kotani K, Kawabe J, Higashiyama S, Yoshida A, Shiomi S (2016) Diffuse Gallium-67 Accumulation in the Left Atrial Wall Detected Using SPECT/CT Fusion Images. Case Rep Radiol 2016: 6374584. [Crossref]
- O'Connell JB, Henkin RE, Robinson JA, Subramanian R, Scanlon PJ (1984). Gallium-67 imaging in patients with dilated cardiomyopathy and biopsy-proven myocarditis. Circulation, 70: 58-62. [Crossref]
- Andreini D, Pontone G, Pepi M, Ballerini G, Bartorelli AL, et al. (2007). Diagnostic accuracy of multidetector computed tomography coronary angiography in patients with dilated cardiomyopathy. J Am Coll Cardiol. 49: 2044-2050. [Crossref]
- Assomull RG, Prasad SK, Lyne J, Smith G, et al. (2006). Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J Am Coll Cardiol 48: 1977-1985.
- Dec GW, Palacios I, Yasuda T, Fallon JT, Khaw BA, et al. (1990). Antimyosin antibody cardiac imaging: Its role in the diagnosis of myocarditis. J Am Coll Cardiol, 16: 97-104. [Crossref]
- Halliday BP, Gulati A1, Ali A1, Guha K1, Newsome S1, et al. (2017) Association Between Midwall Late Gadolinium Enhancement and Sudden Cardiac Death in Patients With Dilated Cardiomyopathy and Mild and Moderate Left Ventricular Systolic Dysfunction. Circulation 135: 2106-2115. [Crossref]
- Machii M, Satoh H, Shiraki K1, Saotome M1, Urushida T, et al. (2014). Distribution of late gadolinium enhancement in end-stage hypertrophic cardiomyopathy and dilated cardiomyopathy: Differential diagnosis and prediction of cardiac outcome. Magn Reson Imaging., 32: 118-124. [Crossref]
- Perazzolo Marra M, De Lazzari M1, Zorzi A, Migliore F, Zilio F, et al. (2014). Impact of the presence and amount of myocardial fibrosis by cardiac magnetic resonance on arrhythmic outcome and sudden cardiac death in nonischemic dilated cardiomyopathy. Heart Rhythm, 11: 856-863. [Crossref]
- McCrohon JA, Moon JC, Prasad SK, McKenna WJ, Lorenz CH, et al. (2003). Differentiation of heart failure related to dilated cardiomyopathy and coronary artery disease using gadolinium-enhanced cardiovascular magnetic resonance. Circulation, 108: 54-59. [Crossref]
- Pinamonti B, Di Lenarda A, Sinagra G, Camerini F (1993). Restrictive left ventricular filling pattern in dilated cardiomyopathy assessed by Doppler echocardiography: clinical, echocardiographic and hemodynamic correlations and prognostic implications. J Am Coll Cardiol, 22: 808-815. [Crossref]
- Puntmann VO, Voigt T, Chen Z, Mayr M, Karim R, et al. (2013). Native T1 mapping in differentiation of normal myocardium from diffuse disease in hypertrophic and dilated cardiomyopathy. JACC: Cardiovascular Imaging, 6: 475-484. [Crossref]
- Sato Y, Yamada T, Taniguchi R, Nagai K, Makiyama T, et al. (2001). Persistently increased serum concentrations of cardiac troponin T in patients with idiopathic dilated cardiomyopathy are predictive of adverse outcomes. Circulation, 103: 369-374. [Crossref]
- Sharp SM, Sawada SG, Segar DS, Ryan T, Kovacs R, et al. (1994). Dobutamine stress echocardiography: detection of coronary artery disease in patients with dilated cardiomyopathy. J Am Coll Cardiol, 24: 934-939. [Crossref]
- McMurray J, Pfeffer MA (2002) New therapeutic options in congestive heart failure: Part I. Circulation 105: 2099-2106. [Crossref]
- Moss AJ, Zareba W, Hall WJ, Klein H, Wilber DJ, et al. (2002). Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med, 346: 877-883. [Crossref]
- Chung ES, Leon AR, Tavazzi L, Sun JP, Nihoyannopoulos P, et al. (2008) Results of the Predictors of Response to CRT (PROSPECT) trial. Circulation 117: 2608-2616. [Crossref]
- Davis J, Davis LC, Correll RN, Makarewich CA4, Schwanekamp JA, et al. (2016). A tension-based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell, 165: 1147-1159. [Crossref]
- Jadad AR, Moore RA, Carroll D, Jenkinson C, Reynolds DJ, et al. (1996) Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Control Clin Trials. 17:1-2. [Crossref]
- Defibrillators, A. V. I. (1997). Investigators. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N Engl J Med 337: 1576-1583. [Crossref]
- Kuck KH, Cappato R, Siebels J, Rüppel R (2000). Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest: the Cardiac Arrest Study Hamburg (CASH). Circulation, 102: 748-754. [Crossref]
- Connolly SJ, Gent M, Roberts RS, Dorian P, Roy D, et al. (2000). Canadian implantable defibrillator study (CIDS): a randomized trial of the implantable cardioverter defibrillator against amiodarone. Circulation, 101: 1297-1302. [Crossref]
- Bänsch D, Antz M, Boczor S, Volkmer M, Tebbenjohanns J, et al. (2002). Primary prevention of sudden cardiac death in idiopathic dilated cardiomyopathy: the Cardiomyopathy Trial (CAT). Circulation, 105: 1453-1458. [Crossref]
- Strickberger SA, Hummel JD, Bartlett TG, Frumin HI, Schuger CD, et al. (2003). Amiodarone versus implantable cardioverter-defibrillator: randomized trial in patients with nonischemicdilated cardiomyopathy and asymptomaticnonsustained ventricular tachycardia—AMIOVIRT. J Am Coll Cardiol, 41: 1707-1712. [Crossref]
- Kadish A, Dyer A, Daubert JP, Quigg R, Estes NA, et al. (2004). Prophylactic defibrillator implantation in patients with nonischemic dilated cardiomyopathy. N Engl J Med, 350: 2151-2158. [Crossref]
- Bardy GH, Lee KL, Mark DB (2004). SCD-HeFT: the sudden cardiac death in heart failure trial. Late Breaking Clinical Trials Session of the 53rd Annual Scientific Sessions of the American College of Cardiology, New Orleans.
- Bristow MR, Saxon LA, Boehmer J, Krueger S, Kass DA, et al. (2004). Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med, 350: 2140-2150.