Abstract
The ventrobasal (VB) thalamus relay nucleus processes information from rodents’ whiskers, projecting to somatosensory cortex. Cocaine and methylphenidate (MPH) have been described to differentially alter intrinsic properties of, and spontaneous GABAergic input to, VB neurons. Here we studied using bis-fura 2 ratiometric fluorescence the effects of cocaine and MPH on intracellular [Ca2+] dynamics at the soma and dendrites of VB neurons.
Cocaine increased baseline fluorescence in VB somatic and dendritic compartments. Peak fluorescence amplitudes and areas were reduced by cocaine binge treatment in somas and dendrites at different holding potentials. MPH binge treatment did not alter ratiometric fluorescence at either somatic or dendritic levels. These novel cocaine-mediated blunting effects on intracellular [Ca2+] might account for alterations in the capacity of thalamocortical neurons to maintain gamma band oscillations, as well as their ability to integrate synaptic afferents.
Key words
Bis-fura 2, cocaine, methylphenidate, thalamocortical, ventrobasal
Abbreviations:
5-HT, serotonin, ADHD: Attention Deficit/Hyperactivity Disorder; DA: Dopamine; DAT: Dopamine Transporter; MPH: Methylphenidate; NE: Norepinephrine; NET: Norepinephrine Transporter; SERT: Serotonin Transporter; VB: Ventrobasal Nucleus
Introduction
Somatosensory thalamus and cortex are massively interconnected [1,2]. The somatosensory thalamus is populated by cortically projecting relay Ventrobasal (VB) neurons that sub serve the thalamocortical loop via glutamatergic synapses onto middle layers of the somatosensory cortex [3]. VB thalamic neurons can display two mutually exclusive electrophysiological modes due to differential calcium channel activation: 1) a tonic-firing mode regulated by subthreshold gamma-band oscillations after thalamocortical neurons are depolarized, which is mediated by the activation of high-voltage activated P/Q-type calcium channels located in VB dendrites [4,5] , or 2) a burst-firing mode when thalamocortical neurons are hyperpolarized, regulated by T-type calcium channel activation [6,7]. Thalamic relay neurons in tonic-firing mode discharge in direct relation to afferent sensory input, whereas, in burst-firing mode, sensory information is not transmitted effectively through thalamocortical networks [8].
Calcium-sensitive dye imaging techniques have been critical in describing how evoked [Ca2+] transients in thalamocortical dendrites can actively propagate and sustain both low and high frequency activity [4,9-12]. Dendrites are key elements for the integration of cortical glutamatergic afferents to VB neurons [13]. Combining high-speed calcium imaging and patch clamp recordings has shown that dendrites from non-specific parafascicular thalamic nucleus can sustain calcium-channels mediated gamma oscillations, at dendritic compartments hundreds of micrometers away from the soma [14]. In specific VB neurons, peak [Ca2+] transient ratios exhibit uniform amplitude along proximo-distal dendritic compartments [9], suggesting the existence of fine intracellular [Ca2+] buffering processes throughout thalamocortical neurons.
Both methylphenidate (MPH) and cocaine have been shown to share mechanistic similarities regarding their ability to increase monoamine concentrations in the synaptic cleft. Indeed, cocaine inhibits dopamine, norepinephrine and serotonin transporters (DAT, NET and SERT, respectively; [15-17]), while MPH mainly inhibits DAT and NET, but not SERT [18,19]. Cocaine abuse is associated with major neuropsychiatric pathologies [20]. MPH is widely used to treat children and adolescents diagnosed with attention deficit/hyperactivity disorder (ADHD; [21]), and has reinforcing effects after intravenous administration in humans [22]. Monoamines like dopamine (DA), norepinephrine (NE), and serotonin (5-HT), released by ascending afferents from brainstem nuclei onto thalamocortical neurons, play an important role in the transitions from low frequency oscillations to high-frequency, gamma band resonance during conscious states [23]. In the somatosensory VB thalamus, cocaine is known to rapidly increase synaptic NE and 5-HT [24], while MPH is able to increase DA levels in the thalamic reticular nucleus (i.e., origin of GABAergic afferents to VB neurons, [25]). A recent study using transgenic mice has described a specific 5-HT modulatory role on GABA release onto VB neurons, modulated by a cocaine binge [26]. Using transgenic mice bypassed previously described collateral effects of SERT inhibitors like fluoxetine on voltage-gated calcium channels [27,28].
Acute binge-like administration of cocaine has been recently shown to alter intrinsic VB neurons’ membrane properties and spontaneous GABAergic inputs to them, resulting in an enhancement of low EEG frequencies [29], through a mechanism that required the over-activation of T-type calcium channels at the level of VB neurons [30]. No clear effects on calcium channels of VB neurons were observed after acute MPH binge-like administration [31]. Although VB neurons can be considered key neurons to understand cocaine and MPH differential effects on thalamocortical networks, little is known about how cocaine and MPH administration can affect their intracellular [Ca2+] dynamics.
We hypothesized that cocaine would induce deeper intracellular [Ca2+] alterations than MPH in VB neurons. We tested [Ca2+]i dynamics using fluorescence ratiometric recordings of thalamic VB neurons from mice treated with either cocaine or MPH binge administration. We used the low/medium affinity dye bis-fura 2 in a patch-clamp whole-cell protocol that differentially activated either T-type or P/Q-type voltage-gated calcium channels in VB neurons of mice treated with cocaine or MPH binge. We found that cocaine, but not MPH, reduced both the peak amplitude and time-integral (i.e., area) of [Ca2+]i signals. Cocaine-mediated alterations of intracellular [Ca2+]i dynamics might have profound consequences on the ability of the dendrites of thalamocortical neurons to integrate synaptic inputs.
Materials and methods
Animals
Preadolescent (18-21 days old) male C57BL/6 mice from the Central Animal Facility at Universidad de Buenos Aires (Animal protocol #50-2015) were used for this study. Principles of animal care were in accordance with “Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research” (National Research Council, 2003), and approved by Universidad de Buenos Aires authorities using OLAW and ARENA directives (NIH, Bethesda, USA).
Drug administration
Cocaine hydrochloride and methylphenidate hydrochloride “binge-like” protocols were i.p. administered (3 injections, 15 mg/kg each, 1 hour apart; [26,29-32]). Control groups received saline injections similarly timed. Animals were sacrificed one hour after the last injection.
Thalamocortical slices
One hour after last i.p. administration, mice were anesthetized with tribromoethanol (250 mg/kg i.p.) followed by transcardial perfusion with ice-cold low sodium/antioxidants solution, and then decapitated. Thalamocortical slices, including the somatosensory cortex (250-350 µm) were obtained using a stage with 55° vertical inclination as previously described [26,29-31]. Slices were then allowed to recover at 35ºC for at least 30 min in a psychostimulant-free ACSF solution.
Whole-cell patch-clamp recordings
Recordings were made in whole-cell voltage-clamp configuration using a MultiClamp 700 amplifier in combination with the pCLAMP 10.0 software (Molecular Devices, CA, USA) at room temperature (20-24°C). Data were filtered at 2 kHz, digitized, and stored for off-line analysis. Patch electrodes were made from borosilicate glass filled with a high Cs+/QX-314 solution (2-5 MW, containing neither EGTA nor CaCl2. Pipettes also contained the low-affinity ratiometric Ca2+-sensitive indicator Bis-fura 2 (Final intracellular concentration 100 µM; stock solutions: 50 mM in DMSO, 1:500 ratio DMSO vs intracellular saline solution). Single 500 ms square pulses were used to evoke voltage dependent calcium currents, changing from a -70 mV holding potential to -30 mV step voltage (i.e., to generate maximum amplitude of mibefradil and 2-octanol sensitive T-type mediated currents; [26,29-31]), or to -10 mV step voltage (i.e., to generate maximum amplitude of ɷ-agatoxin-IVA sensitive P/Q-type currents; [30,31]). Pulses to -30 mV always preceded pulses to -10 mV.
Ratiometric calcium imaging
Ca2+-imaging was performed concomitantly with whole-cell recordings using an Andor iXon+ EMCCD camera (512 x 512, Andor tech., Belfast, UK) coupled to a monochromator Polychrome V (TILL Photonics GmbH, Munich, Germany) through an optic fiber connected to a BX51WI Olympus upright microscope (Olympus Latin America Inc., FL, USA). To allow sufficient filling of dendrites with dye, imaging was initiated 5-10 min after obtaining whole-cell configuration. The low/medium-affinity ratiometric Ca2+-sensitive indicator Bis-fura 2 (in vitro Kd =0.37 µM was previously described in #33) was chosen in order to allow a more accurate report of the time course and amplitude of intracellular [Ca2+] dynamics [14,34]. The experimental protocol is shown in Figure 1A. Baseline ratios were obtained during the 5 s prior to the application of the square voltage pulses while holding membrane potential at -70 mV. The voltage was then stepped to -30 or to -10 mV and fluorescence peak and areas were measured for 16 s after the end of the pulses. Fluorescence changes at 510 nm were acquired after consecutive 340 nm and 380 nm wavelengths using a 0.8 numerical aperture water-immersion 40X objective (Olympus Latin America Inc., FL, USA). Image acquisition was performed using cell-R software (Olympus Soft Imaging Solutions, Münster, Germany), and analyzed post hoc using NIH Image J software. Changes in fluorescence ratios [F(Ratio)=F340/F380] were calculated offline, as the change in fluorescence from baseline of the Ca2+-sensitive indicator bis-fura 2. Baseline fluorescence ratio was calculated by averaging 5 s of fluorescence signal before the square pulse voltage protocol was applied. No direct correlation was obtained between baseline levels and voltage step mediated fluorescence responses. Exposure time to each wavelength was 10 ms and the sampling rate was 10 Hz.
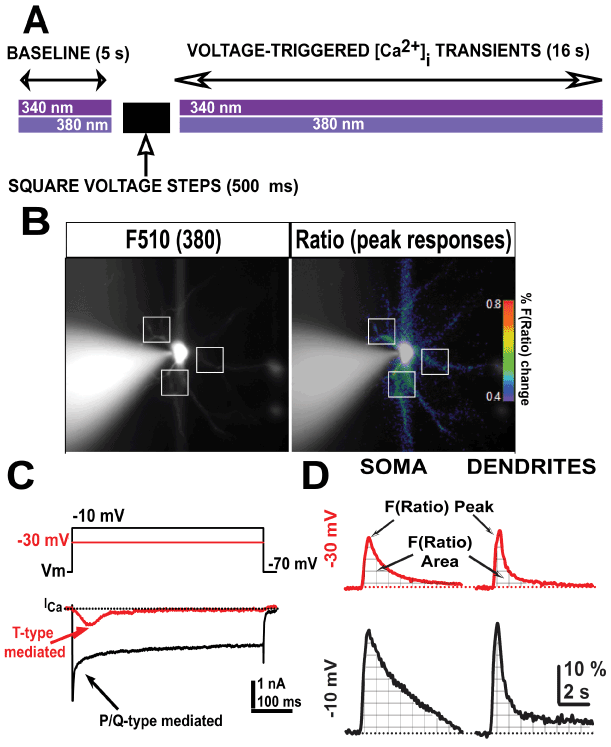
Figure 1. Schematic protocol representation of both ratiometric [Ca2+] fluorescence and voltage-clamp recordings. A, schematic description of the protocol used during consecutive imaging and electrophysiological whole-cell voltage-clamp acquisition. B, representative fluorescence emission from a Bis-fura 2-filled Ventrobasal (VB) neuron excited at 380 nm wavelength (left) and after calculating the fluorescence ratio F(Ratio)=F340/F380 (right) at resting holding potential (-70 mV). Representative dendritic regions of interest are indicated (white squares). The pseudo color bar represents the percentage of Bis-fura 2 mediated F(Ratio) shown in right panel. C, representative T-type calcium currents from a VB neuron elicited by 500 ms long square pulses from holding potential -70 mV to -30 mV (red line) or -10 mV (black line), respectively. D, Representative fluorescent ratio (F(Ratio)) recordings from somatic and dendritic compartments obtained from two different VB neurons. Arrows indicate peak amplitudes and areas (together with grid lines) of F(Ratio) fluorescence signals acquired after -30 and -10 mV holding potential 500 ms pulses in VB neurons.
Statistical analysis
InfoStat software (Universidad Nacional de Córdoba, Argentina) was used for statistical comparisons. Differences were considered significant if p<0.05. Population statistics are presented as mean ± standard error of the mean.
Materials
Cocaine-HCl was purchased from Sigma-Aldrich (St. Louis, MO, USA) and methylphenidate-HCl (Mallinckrodt Inc., Hazelwood, MO, USA) was a generous donation from Osmótica Pharmaceuticals S.A. (Buenos Aires, Argentina). Bis-fura 2 dye was purchased from Molecular Probes (Invitrogen, Grand Island, NY, USA). All other drugs were purchased from either Sigma-Aldrich (St. Louis, MO, USA) or Tocris (Ellisville, MO, USA).
Results
We studied intracellular [Ca2+] dynamics evoked by depolarizing square pulses at the somatic and dendritic compartments of VB neurons from mice treated with cocaine or methylphenidate (MPH), compared to saline (control) conditions. In particular, we studied how intracellular [Ca2+] dynamics changed over time after the membrane potential was rapidly depolarized using 500 ms square voltage pulses (Figure 1A).
We used a high Cesium/QX-314 intracellular solution in voltage-clamp configuration to allow for activation of voltage-dependent calcium currents, but not potassium or sodium currents. Ratiometric [Ca2+] indicator bis-fura 2 was added to the intracellular solution and VB neurons were allowed to passively fill after gaining access to the cytoplasm for 5-10 min to reach a steady-state filling of dendritic compartments (Figure 1B, left panel, white squares). Ratiometric changes in fluorescence (F(Ratio)=F340/F380) were calculated offline and represented as percentage of change from baseline. Step voltage pulses (500 ms) designed to maximally activate either T-type (Figure 1C, red trace; from -70 mv to -30 mV), or P/Q-type (Figure 1C, black trace; from -70 mV to -10 mV) voltage-gated calcium currents were applied to VB neurons to then observe the time course of intracellular [Ca2+] dynamics over the next 16 s (Figure 1A).
While a large somatic region of interest was taken to measure the fluorescence time course at the soma (avoiding places were the fluorescence emission was saturating around the recording electrode), fluorescence values from several proximal dendritic regions were combined into a total dendrite value per neuron (Figure 1B, square white pulses).
Figure 1D shows representative F(Ratio) responses to these stimuli at somatic and dendritic compartments in control conditions. To quantify these results, we measured the maximum peak amplitude and the area following the pulse (Figure 1D, arrows and grid lines under fluorescence signals).
Cocaine increased the baseline fluorescence at the soma of VB neurons, but MPH had no effect (Figure 2A; Kruskal-Wallis ANOVA, H=16.6, P<0.001; Dunn’s post hoc test, cocaine vs. saline, MPH, Q=16, P<0.05; MPH vs. saline, cocaine; Q=2.0, P>0.05). At dendritic levels, both cocaine and MPH increased baseline fluorescence compared to saline (Figure 2B; ANOVA F(2,34)=32.5, P<0.001; Bonferroni’s post hoc test, cocaine vs. saline, t=8.1, P<0.001; MPH vs. saline, t=2.9, P<0.05; cocaine vs. MPH, t=4.5, P<0.001).
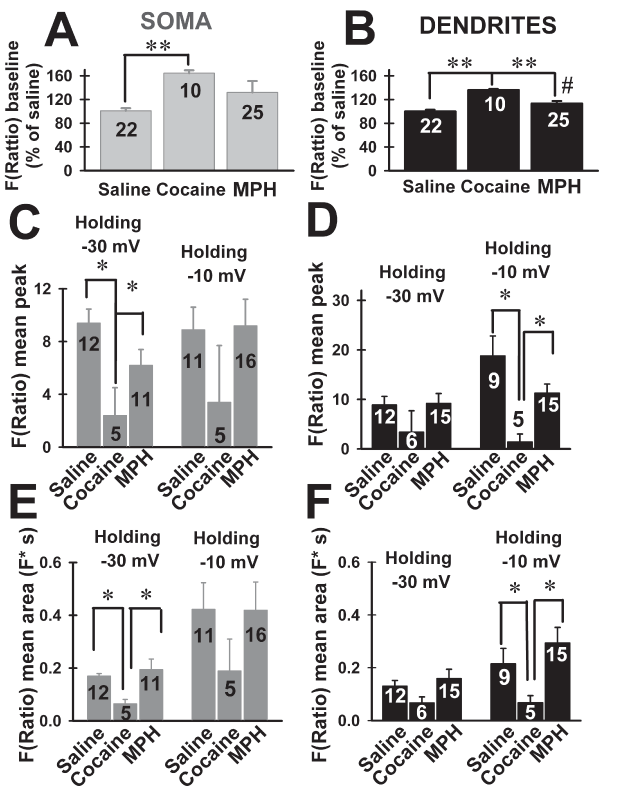
Figure 2. Effects of cocaine and methylphenidate (MPH) binge treatment on peak amplitude and area of F(Ratio) using bis-fura 2 in ventrobasal (VB) neurons. A, B, mean baseline F(Ratio) (shown as percentage of saline values) recorded from soma or dendrites, respectively. ** represents P<0.001 comparing cocaine vs. saline, MPH; # represents P<0.05 comparing MPH vs. saline. C, D, mean peak amplitude F(Ratio) from soma or dendrites, respectively, of VB neurons from mice treated with saline, cocaine or MPH binge treatments. Peak amplitudes were obtained immediately after 500 ms square pulses that changed holding potential from -70 mV to either -30 mV or -10 mV * represents P<0.05. E, F, mean areas (i.e., time-integral of F(Ratio) signals) from soma or dendrites, respectively, of VB neurons from mice treated with saline, cocaine or MPH binge treatments. Peak amplitudes were obtained immediately after 500 ms square pulses that changed holding potential from -70 mV to either -30 mV or -10 mV represents P<0.05.
Peak amplitudes of F(Ratio) signals were compared across treatments at somatic and dendritic compartments (Figure 2C and 2D). Kruskal-Wallis One Way ANOVA showed significantly different peaks at -30 mV across treatments (Figure 2C and 2D; H=15.12, P=0.01). Cocaine decreased the peak F(Ratio) amplitudes measured at -30 mV at the soma (Figure 2C, Kruskal-Wallis ANOVA, H=10.82, P=0.004; Dunn’s pot hoc test; saline vs. cocaine Q=2.86, P<0.05; MPH vs. cocaine Q=2.67, P<0.05). Cocaine and MPH had no significant effect on the F(Ratio) peaks at dendritic levels measured at -30 mV (Figure 2D, Kruskal-Wallis ANOVA, H=3.48, P=0.17).
One way ANOVA comparison of F(Ratio) peak amplitudes also showed significant differences at -10 mV holding potential (Figure 2C and 2D; ANOVA, F(5,64)=4.45, P=0.002). Cocaine, but not MPH reduced peak amplitudes at dendrites (Figure 2D, Bonferroni’s post hoc test, cocaine vs. saline t=3.37, P<0.05; cocaine vs. MPH t=2.8 P<0.05; saline vs. MPH t=0.9 P>0.05), but not at the somas (Figure 2C, P=0.5).
Ratios of peak amplitudes were not significantly different across treatments at any holding potentials (data not shown, Kruskal-Wallis ANOVA, H=4.13, P=0.53). These results suggested that cocaine or MPH induced linear [Ca2+] changes within VB neurons compartments.
Comparison of F(Ratio) areas (i.e., time-integral values) across treatments is shown in Figure 2E and 2F. At the same holding potential, significant changes were observed at both somatic and dendritic compartments. At -30mV, the area measured at the soma was smaller in VB neurons from cocaine-treated mice compared with saline or MPH-treated animals (Figure 2E) (Kruskal Wallis ANOVA, H=7.21, P=0.027; Dunn’s pot hoc test, saline vs. cocaine Q=2.47, P<0.05, MPH vs. cocaine Q=2.46, P<0.05, MPH vs. saline Q=0.08, P>0.05). However, no cocaine effect was observed at the dendrites (ANOVA, F(2,31)=2.79, P=0.08).
At a holding of -10 mV, areas were significantly smaller at dendritic levels after cocaine binge administration (Figure 2F; ANOVA F(2,28)=7.26, P=0.003; Tukey-Kramer post hoc test, cocaine vs. MPH, saline, P<0.05), while remaining non significantly different when comparing cocaine and saline, MPH at the soma (Figure 2E; ANOVA, F(2,32)=1.69, P=0.2). In addition, no increase in area was observed comparing holding potentials at dendritic levels across treatments (Kruskal Wallis ANOVA, P>0.05). Thus, only cocaine binge administration was able to reduce F(Ratio) signals (both peaks and areas) at the somatic or dendritic level at the -30 mV or -10 mV holding potentials, respectively. MPH did not alter F(Ratio) signals at any of the holding potentials tested neither at somatic nor at dendritic levels.
Discussion
These results are the first description of cocaine and MPH binge treatment-mediated [Ca2+] changes in thalamocortical ventrobasal (VB) neurons, suggesting the existence of differential cocaine and MPH effects on intracellular [Ca2+]i dynamics. [Ca2+]i dynamics changed in magnitude (both peak and area) when T-type calcium channels were activated using depolarizing square pulses. In addition, F(Ratio) peaks and areas were smaller after cocaine treatment when P/Q-type calcium channels were activated at the dendrites. For all other conditions, the blunting effect of cocaine was robust, including reduction in peaks and areas of F(Ratio).
Baseline fluorescence levels were higher in VB neurons from cocaine-treated mice, indicating higher levels of [Ca2+]i, in agreement with previously described cocaine-induced T-type calcium channel over- activation closer to resting membrane potentials [29]. Larger baseline [Ca2+] levels would also partially inactivate calcium currents, which might induce lower amplitude [Ca2+]i transients due to its regulation of intracellular pathways [35,36].
Here we also observed a cocaine-mediated reduction in F(Ratio) during T-type calcium channels activation, suggesting the existence of compensatory mechanisms in VB neurons in order to manage higher [Ca2+]i levels through the activation of these channels. Indeed, acute cocaine has been shown to enhance basal [Ca2+]i in cortical neurons [37], while repeated cocaine exposure led to a deregulation of [Ca2+]i homeostasis in nucleus accumbens neurons, that reduced calcium entry [38]. Acute cocaine administration can increase the expression of endoplasmic reticulum stress proteins in striatal neurons [39]. Strikingly, our results suggested that cocaine binge administration did drive VB neurons (neurons of a glutamatergic phenotype) to manage high levels of intracellular [Ca2+], similar to what was described for GABAergic inhibitory neurons [40,41].
A pivotal study by Pedroarena & Llinás [4] described the fact that P/Q-type calcium channels opening at the dendritic compartment of VB neurons are responsible for sustaining gamma band frequency membrane potential sub-threshold oscillations. Dendritic compartments of VB neurons are responsible for integrating cortico-thalamic excitatory inputs [13], creating a strategic condition in which small variations on intracellular [Ca2+] might result in altered thalamocortical dynamics. Simultaneous high-speed video recordings of fura 2-dependent fluorescence and intracellular current clamp recordings showed Ca2+ entry in dendritic compartments in conjunction with injected depolarizing currents via the VB soma [4]. Another report, using voltage-clamp recordings, has shown that largest amplitude of [Ca2+]i signals were observed at proximal dendrites, during a step voltage depolarization capable of activating either T-type or P/Q-type channels [12].
The effects of systemically administered cocaine and MPH on thalamic intracellular [Ca2+] are poorly understood. Thalamocortical VB neurons are known to finely tune intracellular [Ca2+] buffering along proximo/distal dendritic compartments (when recorded in current-clamp mode; [9]). Baseline levels of [Ca2+] ranged from 50-80 nM in parafascicular thalamic neurons using bis-fura 2 [14]. Dendrites can actively sustain low frequency activity in thalamocortical neurons [9-11]. These authors showed dendritic [Ca2+] dynamics (using fluorescence recordings of fluo-filled thalamocortical neurons) during repetitive low frequency spikes [10,11]. Morever, ryanodine receptor-dependent Ca2+-induced Ca2+ release [11,34], and Ca2+-dependent K+ channel activation [42], might blunt the amplitude of [Ca2+] mediated fluorescence signals. However, short depolarizing square pulses used in this work would prevent the activation of intracellular Ca2+-induced Ca2+ release mechanisms [34]. In this sense, recent experiments have shown that [Ca2+]i transients were sensitive to specific toxins against Ca2+ channels, suggesting that Ca2+ entered mainly through voltage-dependent Ca2+ channels in thalamic neurons [14].
Our results also showed clear cocaine-mediated reductions in both peak and area of F(Ratio) after P/Q-type calcium channels were activated in dendrites, probably suggesting a compensatory, faster buffering mechanism in VB dendrites. Future experiments are still needed to further explain cocaine effects on intracellular [Ca2+] reservoirs in thalamocortical neurons.
MPH has not previously been associated with increases in [Ca2+] transients, as shown for cocaine [43]. Du et al. [43] used calcium probes that had no direct relation with the membrane potential of cortical neurons or the type of Ca2+ channels (or other intracellular mechanisms). In spite of that, our results clearly showed strong alterations of [Ca2+] dynamics at VB thalamic neurons after cocaine, but not MPH binge administration, similarly to what Du et al. [43] described at a cortical level.
Describing how intracellular [Ca2+] changes can be influenced by cocaine or MPH is essential to understanding their long-lasting effects on a wide number of the intracellular pathways that use Ca2+ as a second messenger. Results presented in this study specifically suggest that cocaine was able to blunt both somatic and dendritic [Ca2+] integration. MPH did not affect [Ca2+]i dynamics. These novel results provide new insights on how cocaine might deregulate low (i.e., T-type channel-mediated) and high frequency (i.e., P/Q-type channel-mediated) dendritic integration in thalamocortical neurons, affecting the neurons’ ability to integrate corticothalamic synaptic afferents.
Acknowledgment
2021 Copyright OAT. All rights reserv
Dr. Bisagno has been authorized to study drug abuse substances in animal models by A.N.M.A.T. (National Board of Medicine Food and Medical Technology, Ministerio de Salud, Argentina). Methylphenidate was a generous donation from Osmótica Pharmaceuticals S.A. (Buenos Aires, Argentina).
This work was supported by grants from FONCYT-Agencia Nacional de Promoción Científica y Tecnológica (Argentina); Préstamo BID 1728 OC.AR. PICT-2012-1769, PICT 2012-0924, PICT 2015-2594, PICT 2016-1728 and UBACYT 2014-2017 #20120130101305BA. In addition, this work was supported by NIH award R01 NS020246, NIH award P20 GM103425 and P30 GM110702.
Conflict of interest
Authors report no financial conflict of interest, or otherwise, related directly or indirectly to this study.
References
- Steriade M, Llinás RR (1988) The functional states of the thalamus and the associated neuronal interplay. Physiol Rev 68: 649-742. [Crossref]
- Llinás RR, Leznik E, Urbano FJ (2002) Temporal binding via cortical coincidence detection of specific and nonspecific thalamocortical inputs: a voltage-dependent dye-imaging study in mouse brain slices. Proc Natl Acad Sci USA 99: 449-454. [Crossref]
- Land PW, Buffer SA Jr, Yaskosky JD (1995) Barreloids in adult rat thalamus: three-dimensional architecture and relationship to somatosensory cortical barrels. J Comp Neurol 355: 573-588. [Crossref]
- Pedroarena C, Llinás R (1997) Dendritic calcium conductances generate high-frequency oscillation in thalamocortical neurons. Proc Natl Acad Sci USA 94: 724-728. [Crossref]
- Rhodes PA, Llinás R (2005) A model of thalamocortical relay cells. J Physiol 565: 765-781. [Crossref]
- Jahnsen H, Llinás R (1984) Electrophysiological properties of guinea-pig thalamic neurones: an in vitro study. J Physiol 349: 205-226. [Crossref]
- Jahnsen H, Llinás R (1984b) Ionic basis for the electro-responsiveness and oscillatory properties of guinea-pig thalamic neurones in vitro. J Physiol (Lond) 349: 227-247.
- McCormick DA, Feeser HR (1990) Functional implications of burst firing and single spike activity in lateral geniculate relay neurons. Neuroscience 39: 103-113. [Crossref]
- Crandall SR, Govindaiah G, Cox CL (2010) Low-threshold Ca2+ current amplifies distal dendritic signaling in thalamic reticular neurons. J Neurosci 30: 15419-15429. [Crossref]
- Errington AC, Hughes SW, Crunelli V (2012) Rhythmic dendritic Ca2+ oscillations in thalamocortical neurons during slow non-REM sleep-related activity in vitro. J Physiol (Lond) 590: 3691-3700.
- Errington AC, Renger JJ, Uebele VN, Crunelli V (2010) State-dependent firing determines intrinsic dendritic Ca2+ signaling in thalamocortical neurons. J Neurosci 30: 14843-14853. [Crossref]
- Zhou Q, Godwin DW, O'Malley DM, Adams PR (1997) Visualization of calcium influx through channels that shape the burst and tonic firing modes of thalamic relay cells. J Neurophysiol 77: 2816-2825. [Crossref]
- Liu XB, Honda CN, Jones EG (1995) Distribution of four types of synapse on physiologically identified relay neurons in the ventral posterior thalamic nucleus of the cat. J Comp Neurol 352:69-91. [Crossref]
- Hyde J, Kezunovic N, Urbano FJ, Garcia-Rill E (2013) Visualization of fast calcium oscillations in the parafascicular nucleus. Pflugers Arch 465: 1327-1340. [Crossref]
- Glowinski J, Axelrod J (1966) Effects of drugs on the disposition of H-3-norepinephrine in the rat brain. Pharmacol Rev 18: 775-785. [Crossref]
- Wise RA, Bozarth MA (1987) A psychomotor stimulant theory of addiction. Psychol Rev 94: 469-492. [Crossref]
- Howes SR, Dalley JW, Morrison CH, Robbins TW, Everitt BJ (2000) Leftward shift in the acquisition of cocaine self-administration in isolation-reared rats: relationship to extracellular levels of dopamine, serotonin and glutamate in the nucleus accumbens and amygdala-striatal FOS expression. Psychopharmacology (Berl) 151: 55-63. [Crossref]
- Kuczenski R, Segal DS (1997) Effects of methylphenidate on extracellular dopamine, serotonin, and norepinephrine: comparison with amphetamine. J Neurochem 68: 2032-2037. [Crossref]
- Pan D, Gatley SJ, Dewey SL, Chen R, Alexoff DA, et al. (1994) Binding of bromine-substituted analogs of methylphenidate to monoamine transporters. Eur J Pharmacol 264: 177-182. [Crossref]
- Devlin RJ, Henry JA (2008) Clinical review: Major consequences of illicit drug consumption. Crit Care 12: 202. [Crossref]
- Biederman J, Wilens T, Mick E, Spencer T, Faraone SV (1999) Pharmacotherapy of attention-deficit/hyperactivity disorder reduces risk for substance use disorder. Pediatrics 104: e20. [Crossref]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, et al. (1999) Reinforcing effects of psychostimulants in humans are associated with increases in brain dopamine and occupancy of D(2) receptors. J Pharmacol Exp Ther 291: 409-415. [Crossref]
- McCormick DA, Bal T (1997) Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci 20: 185-215. [Crossref]
- Rutter JJ, Baumann MH, Waterhouse BD (1998) Systemically administered cocaine alters stimulus-evoked responses of thalamic somatosensory neurons to perithreshold vibrissae stimulation. Brain Res 798: 7-17. [Crossref]
- Erlij D, Acosta-García J, Rojas-Márquez M, González-Hernández B, Escartín-Perez E, et al. (2012) Dopamine D4 receptor stimulation in GABAergic projections of the globus pallidus to the reticular thalamic nucleus and the substantia nigra reticulata of the rat decreases locomotor activity. Neuropharmacology 62: 1111-1118. [Crossref]
- Goitia B, Rivero-Echeto MC, Weisstaub NV, Gingrich JA, Garcia-Rill E, et al. (2016) Modulation of GABA release from the thalamic reticular nucleus by cocaine and caffeine: role of serotonin receptors. J Neurochem 136: 526-535. [Crossref]
- Deák F, Lasztóczi B, Pacher P, Petheö GL, Valéria K, et al. (2000) Inhibition of voltage-gated calcium channels by fluoxetine in rat hippocampal pyramidal cells. Neuropharmacology 39: 1029-1036. [Crossref]
- Traboulsie A, Chemin J, Kupfer E, Nargeot J, Lory P (2006) T-type calcium channels are inhibited by fluoxetine and its metabolite norfluoxetine. Mol Pharmacol 69: 1963-1968. [Crossref]
- Urbano FJ, Bisagno V, Wikinski SI, Uchitel OD, Llinás RR (2009) Cocaine Acute “Binge” Administration Results in Altered Thalamocortical Interactions in Mice. Biol Psychiatry 66: 769-776. [Crossref]
- Bisagno V, Raineri M, Peskin V, Wikinski SI, Uchitel OD, et al. (2010) Effects of T-type calcium channel blockers on cocaine-induced hyperlocomotion and thalamocortical GABAergic abnormalities in mice. Psychopharmacology (Berl) 212: 205-214. [Crossref]
- Goitia B, Raineri M, González LE, Rozas JL, Garcia-Rill E, et al. (2013) Differential effects of methylphenidate and cocaine on GABA transmission in sensory thalamic nuclei. J Neurochem 124: 602-612. [Crossref]
- Schlussman SD, Zhang Y, Kane S, Stewart CL, Ho A, et al. (2003) Locomotion, stereotypy, and dopamine D1 receptors after chronic "binge" cocaine in C57BL/6J and 129/J mice. Pharmacol Biochem Behav 75: 123-131. [Crossref]
- Takahashi A, Camacho P, Lechleiter JD, Herman B (1999) Measurement of intracellular calcium. Physiol Rev 79: 1089-1125. [Crossref]
- Budde T, Sieg F, Braunewell KH, Gundelfinger ED, Pape HC (2000) Ca2+-induced Ca2+ release supports the relay mode of activity in thalamocortical cells. Neuron 26: 483-492. [Crossref]
- Perez-Reyes E (2003) Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev 83: 117-161. [Crossref]
- Mitani K, Sekiguchi F, Maeda T, Tanaka Y, Yoshida S, et al. (2016) The prostaglandin E2/EP4 receptor/cyclic AMP/T-type Ca(2+) channel pathway mediates neuritogenesis in sensory neuron-like ND7/23 cells. J Pharmacol Sci 130(3):177-180. [Crossref]
- Cunha-Oliveira T, Rego AC, Garrido J, Borges F, Macedo T, et al. (2010) Neurotoxicity of heroin-cocaine combinations in rat cortical neurons. Toxicology 276: 11-17. [Crossref]
- Perez MF, Ford KA, Goussakov I, Stutzmann GE, Hu XT (2011) Repeated cocaine exposure decreases dopamine D2-like receptor modulation of Ca(2+) homeostasis in rat nucleus accumbens neurons. Synapse 65: 168-180. [Crossref]
- Shin EH, Bian S, Shim YB, Rahman MA, Chung KT, et al. (2007) Cocaine increases endoplasmic reticulum stress protein expression in striatal neurons. Neuroscience 145: 621-630. [Crossref]
- Fierro L, Llano I (1996) High endogenous calcium buffering in Purkinje cells from rat cerebellar slices. J Physiol (Lond) 496: 617-625. [Crossref]
- Lee SH, Rosenmund C, Schwaller B, Neher E (2000) Differences in Ca2+ buffering properties between excitatory and inhibitory hippocampal neurons from the rat. J Physiol (Lond) 525: 405-418. [Crossref]
- Cueni L, Canepari M, Luján R, Emmenegger Y, Watanabe M, et al. (2008) T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat Neurosci 11: 683-692. [Crossref]
- Du C, Yu M, Volkow ND, Koretsky AP, Fowler JS, et al. (2006) Cocaine increases the intracellular calcium concentration in brain independently of its cerebrovascular effects. J Neurosci 26: 11522-11531. [Crossref]