The current research describes the design, synthesis, and in vitro evaluate as DPPIV inhibitors for four new compounds. The design of the new compounds aimed to study the effect of replacing the 2-benzyl-piperazine ring system of a reported potent DPP IV inhibitor, IC50 of 19 nM, with piperidine and pyrrolidine heterocyclic ring systems. The newly synthesized compounds, along with the lead compound, were studied for docking affinities and mode of interactions with the DPP active site using molecular modelling approach in an attempt to correlate between the binding parameters and the observed in vitro inhibitory activities.
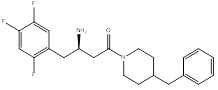
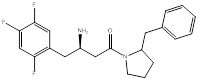
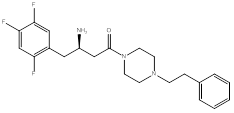
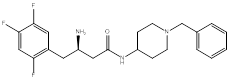
Compound (1), a 4-benzylpiperidine derivative, possessed a ΔG value of -8.9 Kcal/mol, and an IC50 of 1.6 ± 0.04 µM. Compound (2), a 2-benzylpyrrolidine derivative, showed a ΔG value of -9.0 Kcal/mol, and an IC50 of 0.3 ± 0.03 µM. Compound (3), a phenethyl-piperazine derivative, exhibited a ΔG value of -8.9 Kcal/mol, and an IC50 of 1.2 ± 0.04 µM. Compound (4), a 4-amino-1-benzylpiperidine derivative, showed a ΔG value of -8.9 Kcal/mol, and an IC50 of 4 ± 0.08 µM.
The study revealed that compound (2), with reduced ring size into pyrrolidine in place of the lead compound piperazine ring, while retaining the benzyl substitution at the 2-position, resulted in the most active inhibitor among the four newly designed compounds; however, with less inhibitory activity in comparison to the reported 2-benzyl-piperazine lead compound.
DPPIV inhibitors, pyrrolidine heterocyclic ring systems, inhibitory activity
Dipeptidyl peptidase IV (DPP IV) is one of the new targets for potential antidiabetic agents. Its inhibition allows glucagon-like peptide-1 (GLP-1) to survive longer in the plasma and potentiate β cell response with minimum risk of hypoglycemia. DPP IV is a serine protease that cleaves dipeptides from the N-terminus of polypeptides where Ala or Pro are in the penultimate position. While DPP IV has many substrates, GLP-1 and glucose-dependent insulinotropic polypeptide (GIP) have received significant attention due to their potential in the treatment of diabetes. DPP IV is expressed in many organs and tissues including the pancreas, gastrointestinal tract, kidney, lymph nodes sweat glands, and mammary glands. It is mainly found as an integral protein that spans the plasma membranes of endothelial cells of many organs, but it can also be found in a soluble form in the blood plasma [1]. Its primary structure consists of a cytoplasmic tail that contains 6 amino acids followed by a transmembrane domain of 22 amino acids as well as an extracellular domain comprised of 738 amino acids. This enzyme contains the catalytic triad Ser630, Asp708, and His740 [2].
When the substrate peptide binds to the active site of DPP IV, its amino group is recognized by Glu205 and Glu206, then Ser630 of the catalytic triad attacks and cleaves the peptide bond after the penultimate amino acid (P1) [3]. Two main pockets of the active site that are targeted for drug design are the S1 and S2 pocket. The S1 pocket is lined with Tyr631, Val656, Trp659, Tyr662, Tyr666, Val711, and Asn710, while the S2 pocket contains Arg125, Phe357, Arg358, Glu205, Glu206, and Arg669 [2,4] (Figure 1).
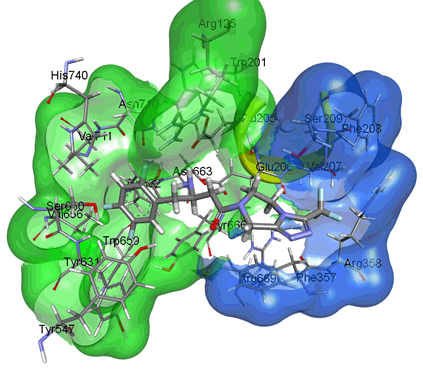
Figure 1. Active sites of DPP IV. The surface map shows S1 pocket in green and S2 pocket in blue. (PDB, ID: 1X70, human DPP IV in complex with sitagliptin. The picture was prepared using the surface tool of the Discovery Studio Visualizer).
Sitagliptin, vildagliptin, saxagliptin, alogliptin, and linagliptin are the first reversible competitive inhibitors to be introduced to the market (Figure 2). The inhibition of DPP IV prevents the degradation of incretins prolonging their half-life. This in turn increases insulin production proportionally to the blood glucose level, thus minimizing the risk of hypoglycemia. The need of alternative therapy to the DPP IV-resistant GLP-1 analogs gave rise to the DPP IV inhibitor series. Sitagliptin, a β-homophenylalanine triazolopiperazine derivative developed by Merck with an IC₅₀ value of 18 nM, possesses two pharmacophores: a β-amino amide and a triazolo piperazine attached to a trifluoromethyl.
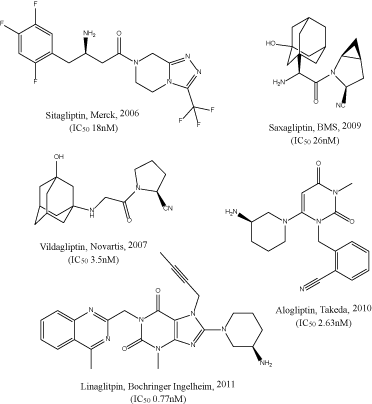
Figure 2. Marketed DPP IV inhibitors
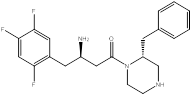
As shown in the figure 2, DPP IV inhibitors do not have a common structural feature, and different SAR studies have been reported [2]. From the different SAR studies of sitagliptin, we noted that a 2-benzylpiperazine derivative had an IC50 of 19nM [5,6] (Figure 3).
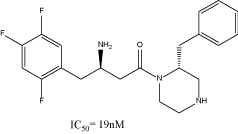
Figure 3. 2-benzylpiperazine derivative, lead compound
Purpose of the study
The research investigated modifications of the piperazine ring in the lead compound (Figure 3), the circled part in figure 4, in attempt to optimize complementary interactions with the amino acid residues of the S2 pocket of DPP IV. The design relied on molecular docking for the interpretation of any observed difference in activity of the proposed compounds with different heterocyclic nuclei and variable positions for the benzyl group. Among the proposed heterocyclic nuclei to replace the piperazine heterocyclic ring were:
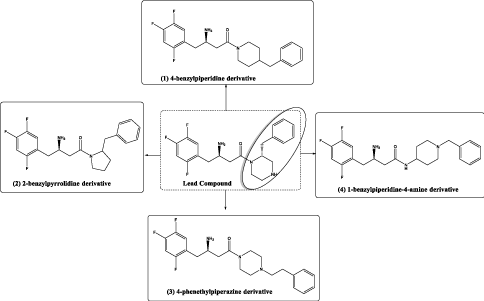
Figure 4. Summary of the design scheme of the compounds reported in this research
1. Piperidine-heterocycle as a bioisostere for piperazine with potential of more hydrophobic interaction, compound (1), (Figure 4).
2. Pyrrolidine-heterocycle to investigate the effect of reducing the ring size into a 5-membered ring, compound (2), (Figure 4).
3. Piperazine-heterocycle with phenethyl group instead of the benzyl to study the effect of side chain extension in interacting with extra hydrophobic amino acid residues, compound (3), (Figure 4).
4. Another piperidine ring system with 4-amino group as a representative for a piperazine- like system, but with one of the nitrogen atoms outside the ring in attempt to introduce structural flexibility [7], compound (4), (Figure 4).
Figure 4 depicts summary of the proposed structures investigated in this research.
All four compounds were synthesized according to standard synthetic schemes and docked in the active site using molecular modelling to determine the binding affinity and the binding mode in the enzyme active site. The research also aims to perform in-vitro testing for the synthesized compound and determine the inhibitory IC50 value for each compound and correlate it with the docking profile.
To test the activity of the new inhibitors, we used the same biochemical assay as described in the literature [5]. The assay was validated by running Michaelis-Menten kinetics and comparing the Km of the substrate with that in the literature.
The assay used is a kinetic fluorometric assay that relies on the fluorogenic substrate Gly-Pro-4-methylcoumarinamide which gets cleaved by DPP-IV enzyme releasing the fluorescent 7-amino-4-methylcoumarin from the C-terminus of the peptide (Figure 5).
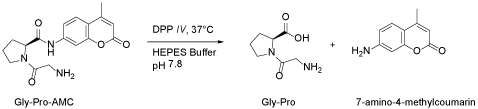
Figure 5. DPP-IV enzyme releasing the fluorescent 7-amino-4-methylcoumarin from the C-terminus of the peptide
The docking profile of the lead compound exhibits complementary interactions of the benzylpiperazine part with the S1 pocket and the β-homophenylalanine part with the S2 pocket. This observation can be used to interpret why the compounds under investigation that exhibit a flipped over orientation compared to lead compound have lower inhibitory activity (Figure 6).
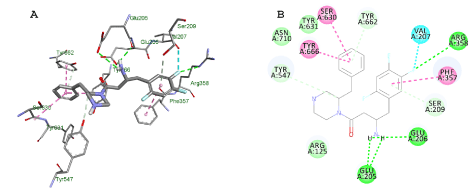
Figure 6. Docking results of 2-benzylpiperazine derivative, lead compound, in the DPP IV active site. Lowest energy pose from the docking studies of the lead compound in the enzyme active site is shown in A and B in 3D and 2D, respectively
Compound (1)
The changes that were investigated, in compound (1), are piperazine replacement by piperidine and benzyl substituent at position 4 instead of 2. The docking result demonstrated a flipped over orientation compared to the lead in the active site binding. The benzyl group became oriented outside the S2 pocket maintaining a weak, but essential interaction with the S2 binding pocket (Figure 7).
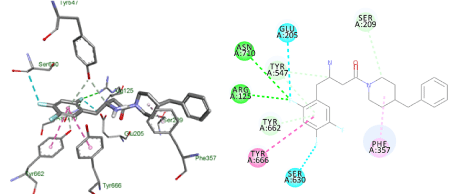
Figure 7. Docking result of compound (1)
Lowest energy poses from the docking studies of compound (1) in the enzyme active site.
Docking studies of compound (1) showed a flipped over orientation compared to the lead with the β-homophenylalanine moiety embedded in the S1 pocket and its ring interacting weakly with Phe357 and Ser209 of the S2 pocket. This can be used to interpret the inferior IC50 value of 1.6 ± 0.04 µM (Figure 8).
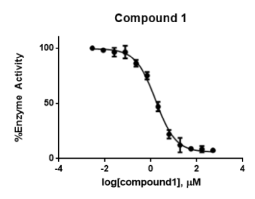
Figure 8. Concentration-response curve of compound (1) Data points represent % Activity of the enzyme ± SD. The data points represent the mean of 3 independent experiments run in triplicates
Compound (2)
The next compound had a reduced ring size with the benzyl group position unchanged. It exhibited a superimposable pose to the lead compound in the active site. This oriented the amino group to interact with both Glu205 and Glu206 residues and maintained the hydrophobic interactions with Phe357, and a weak hydrogen bond to Ser209 (Figure 9).
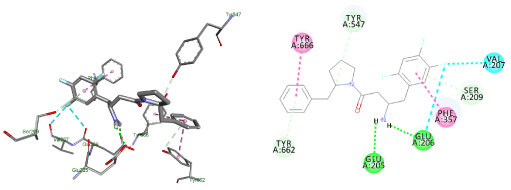
Figure 9. Docking result of compound (2)
Lowest energy poses from the docking studies of compound (2) in the enzyme active site.
Compound (2) was superimposed with the lead compound and maintained essential interactions with the amino acid residues of S2 pocket. This explains the IC50 of 0.3 ± 0.03 μM and suggests that a benzyl substituent at position 2 has more potent inhibitory activity (Figure 10).
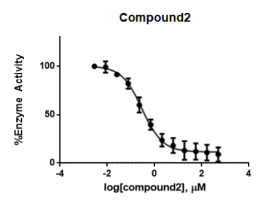
Figure 10. Concentration-response curve of compound (2) Data points represent % Activity of the enzyme ± SD. The data points represent the mean of 3 independent experiments run in triplicates
Compound (3)
The next compound had a piperazine ring system and the benzyl substituent at position 4 with extension of the side chain to target interaction with Arg358. The binding exhibited a flipped over orientation compared to that of the lead and lacked interaction with Glu205, Glu206, and Ser209. The extension of the side chain dictated that the lowest binding pose be in a manner that hindered complementary interactions of the amino groups with the pocket residues (Figure 11).
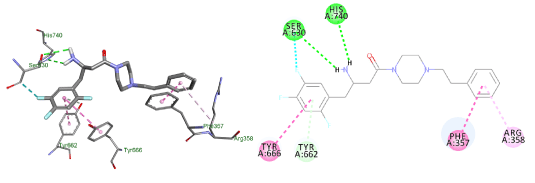
Figure 11. Docking result of compound (3)
Lowest energy poses from the docking studies of compound (3) in the enzyme active site.
In the enzymatic assay, compound (3) shows an IC50 value of 1.2 ± 0.04 μM. This result suggests that adding an extra carbon spacer, and probably the change of substituent position as well, lowers the activity (Figure 12).
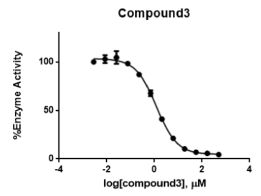
Figure 12. Concentration-response curve of compound (3) Data points represent % Activity of the enzyme ± SD. The data points represent the mean of 3 independent experiments run in triplicates.
Compound (4)
The last compound had a piperidine ring with the amide group outside the ring system to increase the flexibility of the molecule. The benzyl substituent at position 4 to target extra hydrophobic interactions with Arg356. The docking exhibited a flipped over pose to the lead and lacked interactions with Glu205 and Ser209 (Figure 13).
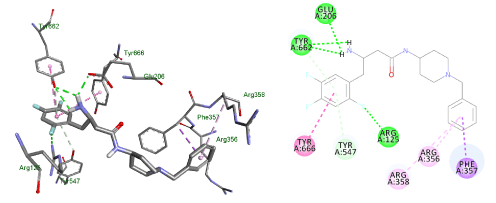
Figure 13. Docking result of compound (4)
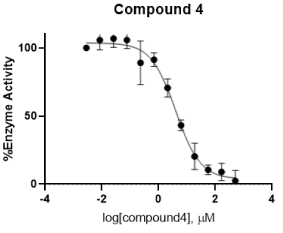
Figure 14. Concentration-response curve of compound (4)
Lowest energy poses from the docking studies of compound (4) in the enzyme active site.
In the enzymatic assay, compound (4) shows an IC50 value of 4 ± 0.08 μM. This result suggests that having the amide group outside the ring system, and probably the change of substituent to position 4 as well, lowers the activity (Figure 13).
Data points represent % Activity of the enzyme ± SD. The data points represent the mean of 3 independent experiments run in triplicates (Table 1).
Table 1. Summary of the IC50 values and the ΔG of the lowest energy pose in the docking study.* Literature-reported IC50 value for the lead compound.6
In this research, the balance between flexibility and charge distribution was investigated in the designed compounds. The data suggests that the optimum inhibitory activity could be achieved in a compound that has the following features: A diheterocyclic ring system, a reduced ring size and a hydrophobic substituent at the 2-position.
The out-of-pocket interaction of the 4-substituent, in compound (1), suggests the more appropriate use of 2-substituent or introducing single bond rotation to increase the flexibility of the inhibitor in the enzyme active site. Compound (2) was the most active inhibitor, superimposing the lead compound in the active site and maintaining all essential complementary interactions with the S2 amino acid residues. This suggests that retaining the substituent at the 2-position is essential for potent inhibitory activity. Extension of the side chain in an attempt to target extra hydrophobic interactions, in compound (3), although giving an active inhibitor, did not show an optimum inhibitory profile. The activity could be contributed to the piperazine ring system, compared to a piperidine one. Extracting the amide group outside the ring system to increase structural flexibility, in compound (4), although retained most essential complementary interactions with the S2 pocket residues, was the least active inhibitor. This suggests that increasing the system’s flexibility lowers the inhibitory activity.
The use of binding affinity to compare the inhibitory activities of the four compounds was inefficacious; the ΔG values did not match the inhibitory activity. Therefore, the determinant factor of activity, besides the IC50 value, was the binding pose and not the binding affinity.
Docking studies
All the docking studies were performed using PyRx (V.0.8) which provides a Microsoft Windows interface for Vina, which is the main docking platform [8,9]. To perform the docking studies, the crystal structure of DPP IV was downloaded in .pdb format (PDB ID 1x70) from the protein databank (www.rcsb.org). To prepare the protein for docking, the .pdb file was imported to Autodock Tools (v.1.5.6). The water molecules and ligands were removed from the structure, the polar hydrogens were added to the residues, then the structure was converted to. pdbqt format in which the partial charges on the residues were calculated. To prepare the ligands for docking, the structure of the ligands was drawn using Marvin Sketch (V.15.11.9) and saved as .sdf format [10]. The protein as well as the ligands files were then imported to PyRx. The energy of ligands were minimized using Open Babel extension in the program, then the files of the ligands were converted to pdbqt format [11]. The grid box for the docking was set to be 25 A° X 25 A° X and was centered on the active site which was located at the following coordinates: X=40.1242, Y=50.9588, Z=36.1963. The docking studies were performed three times and the potential energies were reported as average of the three runs. The docking files were then imported to Discovery Studio Visualizer (V. 4.5.0.15071) which was used to visualize the interactions represented in the figures [12].
Synthesis
Reagents were used as received from commercial suppliers, Sigma Aldrich and aa Blocks. Melting points of the compound was taken using DigiMelt MPA160. 1H-NMR was recorded at 300 MHz on a Varian instrument using VnmrJ version 4.2A. Elemental analyses were performed by Atlantic Microlab Inc., Norcross, GA. All MS experiments were carried out on an AB/Sciex 4000 LC/MS/MS triple quadrupole using electrospray ionization in positive ion mode. Flash chromatography was performed on a CombiFlash NEXTGEN 100 using a 12-gram HP silica column supplied from Teledyne ISCO. For biological testing, a fluorometric assay was performed in Corning 96 well black plates with opaque bottom on a Synergy Bio-tek HTS plate reader with excitation/ emission filters of 360/460 nm using Gen5 software. All the buffers used for the assay were prepared in the lab according to the procedure described in the literature [13].
tert-butyl(R)-(4-(4-benzylpiperidin-1-yl)-4-oxo-1-(2,4,5-trifluorophenyl)butan-2-yl)carbamate (1)
To a 50 mL round bottom flask (RBF) the Boc-protected amine was added 2.00 g (6.00 mmol) followed by 20 mL of dry THF and 1.89g of DEPBT (6.30 mmol). The purpose of this step is to activate the Boc-protected amine for condensation with the 4-benzyl piperidine. The activation step was followed by TLC (50% ethyl acetate/hexane- Rƒ 0.66) and completion detected after 1 hour. To the solution, 4-benzyl piperidine 1.1 g (6.30 mmol) mixed with TEA 2.5 mL (6.00 mmol) and THF 7 mL was added. The reaction was left to stir for 12 hours at room temp, then the solvent was evaporated in-vacuo. The residue left was dissolved in ethyl acetate and was washed with 1M HCl (45 mL) twice, saturated solution of NaHCO₃ (45 mL) thrice, and distilled water (45 mL) once, then brine (45 mL) thrice. The organic solvent was then dried over anhydrous sodium sulfate, filtered then evaporated, and the remaining residue was dissolved in minimum amount of ethyl acetate and allowed to crystallize in hexane over ice to afford 1.99 g of pure white crystals (67.7% yield) with m.p. 135-136°C. 1H-NMR (300 MHz, DMSO-d6) δ 7.3 (s, 1H), 7.28 (m, 5H), 7.2 (m, 1H), 7.12 (m, 1H), 4.16 (p, J=7.0 Hz, 1H), 3.72 (dt, J=7.1 Hz, 4H), 2.5-2.9 (qd, J=7.0 Hz, 4H), 2.0 (m, 2H), 1.6 (m, 1H), 1.25 (m, 3H). MS m/z 491 (M + H)+. To 800 mg of the above intermediate 4 mL of 4N HCl in dioxane was added and the solution was left to stir for 1.5 h before the TLC indicated the completion of the reaction. The precipitate was washed with dry diethyl ether and crystallized using 1:10 methanol/diethyl ether to afford 241 mg of pure white crystals (37.7% yield) with m.p. 144-146°C. 1H-NMR (300 MHz, DMSO-d6) δ 7.98 (s, 3H), 7.56 (m, 5H), 7.26 (t, J=7.5Hz, 1.5 Hz, 1H), 7.18 (t, J=8.0 Hz, 5.0 Hz, 1H), 4.32 (m, 1H), 3.7 (m, 4H), 2.87 (m, 2H), 2.44 (m, 2H), 1.68 (t, J=7.1 Hz, 2H), 1.58 (d, J=7.0 Hz, 1H), 0.98 (m, 4H). Microanalysis: (C, 61.99%), (H, 6.13%), (N, 6.5%), (Cl,8.23%), and (F, 13.39%).
(3R)-3-amino-1-(2-benzylpyrrolidin-1-yl)-4-(2,4,5-trifluorophenyl)butan-1-one (2)
To a 50 mL RBF the Boc-protected amine was added 2.00 g (6.00 mmol) followed by 10 mL of dry THF and 1.89g of DEPBT (6.00 mmol). To the solution, 2-benzylpyrrolidine 1.94 g (12.00 mmol) mixed with 2 mL TEA and 4 mL THF was added and left to stir at 40 °C. The reaction was followed by TLC (70% ethyl acetate/hexane- Rƒ 0.66) and completion detected after 19 hrs followed by solvent evaporation. The residue left was dissolved in ethyl acetate and was washed with HCl (45 mL) twice, sat. solution of NaHCO₃ (45 mL) thrice, and distilled water (45 mL) once, then brine (45 mL) thrice. The organic solvent was then dried over anhydrous sodium sulfate then evaporated, and the remaining residue was dissolved in acetone and crystallized with distilled water three times to afford 0.71 g yellow crystals (yield 63.8%) with m.p. 123-125 °C. 1H NMR (300 MHz, DMSO-d6, mixture of diastereomers) δ 7.32 (s, 1H), 7.3 (m, 5H), 7.12 (m, 1H), 6.9 (m, 1H), 4.4 (m, 1H), 4.18 (m, 1H), 3.3 (m, 2H), 3 (s, 1H), 2.6 (m, 3H), 1.4, 1.8 (m, 4H), 1.1 (s, 1H). Microanalysis: (C, 65.47%), (H, 6.70%), (N, 5.85%), and (F, 12.05%). To 950 mg of the above intermediate 4 mL of 4N HCl in dioxane and the solution was left to stir for 22 h before the TLC indicated the completion of the reaction. The addition of dry diethyl ether gave a yellowish residue, so the free base was obtained by alkalization with NaHCO3 and extraction in chloroform. The chloroform was dried with sodium sulfate and evaporated giving a yellow residue. Purification was performed using flash chromatography by gradient elution with ethyl acetate and hexane, silica 12 g column, and a flow rate of 30 mL/min. The pure fraction was evaporated affording 500 mg of yellow oil (66.6% yield). 1H NMR (300 MHz, DMSO-d6, mixture of diastereomers) δ 7.25 (m, 5H), 7.1(m, 1H), 6.9 (m, 1H), 4.32 (m, 1H), 3.6 (m, 2H), 3.5 (m, 1H), 3.2 (m, 2H), 2.8 (qd, J= 7.0, 2H), 2.6 (m, 2H), 2.4 (m, 2H), 1.74 (s, 1H). MS m/z 377 (M + H)+.
(R)-4-oxo-4-(4-phenethylpiperazin-1-yl)-1-(2,4,5-trifluorophenyl)butan-2-aminium chloride (3)
To a 50 mL RBF the Boc-protected amine was added 3.00 g (9.00 mmol) followed by 15 mL of dry THF and 2.96 g of DEPBT (9.00 mmol). To the solution, 1-phenethylpiperazine HCl 1.88 g (12.00 mmol) mixed with 3 mL TEA (27.00 mmol) and 5 mL THF was added and left to stir at room temp. The reaction was followed by TLC (50% ethyl acetate/hexane- Rƒ 0.2) and completion detected after 24 hrs followed by solvent evaporation. The residue left was dissolved in ethyl acetate and was washed with HCl (45 mL) twice, sat. sol of NaHCO₃ (45 mL) thrice, and distilled water (45 mL) once, then brine (45 mL) thrice. The organic solvent was then dried over anhydrous sodium sulfate, filtered, then evaporated, and the remaining residue was dissolved in acetone and crystallized with distilled water three times to afford 3 g white crystals (yield 77.9%) with m.p. 123-126 °C. 1H NMR (300 MHz, CDCl3) δ 7.32 (s, 1H), 7.25 (m, 5H), 7.1 (m, 1H), 6.9 (m, 1H), 4.1 (p, J=7.0, 1H), 3.6 (m, 4H), 2.9 (m, 2H), 2.82 (t, J= 7.1, 2H) 2.55 (qd, J= 7.1 Hz, 2H), 2.2 (s, 9H), 1.2 (s, 9H). MS m/z 506 (M + H)+. To the above 2.18 g of the intermediate 7 mL of 4N HCl in dioxane and the solution was left to stir for 1 h before the TLC indicated the completion of the reaction. The formed precipitate was washed with dry diethyl ether and crystallized using methanol/diethyl ether to afford 1.65 g of pure white crystals (84.5% yield) with m.p. 248-250 °C. 1H NMR (300 MHz, DMSO-d6) δ11.9 (s, 1H), 8.35 (d, J=7.1 Hz, 3H), 7.61 (d, J= 7.5 Hz, 3H), 7.3 (m, 5H), 4.41 (m, 1H), 3.99 (d, J= 7.1 Hz, 1H), 3.72 (t, J= 7.0 Hz, 2H), 3.56 (s, 10H), 3.16 (m, 1H), 2.9 (m, 1H). Microanalysis: (C, 54.59%), (H, 6.06%), (N, 8.66%), (Cl, 14.5%), and (F, 11.68%).
(R)-3-amino-N-(1-benzylpiperidin-4-yl)-4-(2,4,5-trifluorophenyl)butanamide (4)
To a 100 mL RBF the Boc-protected amine was added 5.00 g (15.0 mmol) followed by 50 mL of dry THF and 4.71 g of DEPBT (15.8 mmol). To the solution, 1-piperidine-4-amine 2.85g (15.0 mmol) mixed with 2.30 mL TEA (16.5 mmol) and 5 mL THF was added and left to stir at room temp in argon gas. The reaction was followed by TLC (100% methanol- Rƒ 0.72) and completion detected after 3 hrs followed by solvent evaporation. Water was added to the residue and the oily material was separated from the solid by decantation. The solid was then washed with a sat. sol of NaHCO₃ (20 mL) four times and distilled water (20 mL) four times. The precipitate was left to dry over vacuum and crystallization was performed by 1:1 DMSO/distilled water twice to yield 4.0g of pure white crystals (52.7% yield) with m.p. 201-202°C. 1H NMR (300 MHz, CDCl3) δ7.73 (d, J= 7.1 Hz, 1H), 7.43 (s, 1H), 7.27 (m, 5H), 6.69 (d, J= 8.0 Hz, 1H), 3.96 (s, 1H), 3.4 (s, 3H), 2.7 (d, J= 7.0 Hz, 2H), 2.23 (dd, J= 7.0, 7.1 Hz, 2H), 1.97 (m, 2H), 1.64 (s, 2H), 1.34 (m, 2H), 1.25 (s, 9H). Microanalysis: (C, 63.87%), (H, 6.82%), (N, 8.17) and (F, 10.99%). To 0.7 g of the above intermediate 5 mL of 4N HCl in dioxane was added followed by 5 mL of 1,4-dioxane. The reaction was left to stir for 18 h before the TLC indicated the completion of the reaction (100% methanol- Rf 0.39). Excess solvent was evaporated, and the formed precipitate was washed with dry diethyl ether and ethyl acetate (10 mL) three times each. White crystals weighing 0.6g were obtained (yield 87.6%) with decomposition that started at 243°C. Different crystallization methods gave an oily residue therefore the product was converted to the free base form. The solid was dissolved in 2 mL of distilled water and pH adjusted to 11 by adding 1N NaOH. The free base was extracted by dichloromethane, the organic layer was then dried with anhydrous sodium sulfate and evaporated in-vacuo. Different crystallization attempts to obtain the product in solid state gave a clear oily residue. Therefore, the purity and identity of the product were confirmed by TLC, H-NMR and MS. 1H NMR (300 MHz, DMSO-d6) δ7.9 (s, 1H), 7.43 (m, 2H), 7.26 (m, 5H), 3.54 (s, 2H), 3.42 (m, 1H), 3.22 (m, 1H), 2.72 (s, 2H), 2 (m, 6H), 1.36 (m, 6H). MS m/z 406 (M + H)+.
Enzyme Eliquots
The enzyme was purchased from Millipore Sigma (Catalog # D3446-10UG) and 100ml of the 800pM stock solution was prepared in 10 mM Tris buffer at pH 8 containing 200 mM NaCl, 1mM EDTA and 10% Glycerol. The enzyme was stored at -70 °C as recommended by the manufacturer. Before performing the biological assay, the enzyme was diluted 4 folds using 100 mM HEPES buffer with pH 7.8 and 1 µg/ml BSA to make an initial enzyme concentration of 200 pM.
Substrate
The substrate was purchased from Millipore Sigma (Catalog# G2761-25MG) and a 16 mM stock solution was prepared by adding 3.81 ml of HEPES buffer to the 25 mg of the peptide.
Assay: Each well except for the blank contained 25 µl of the enzyme, 25 µl of the substrate, and 50 µl of the test compounds bringing the final enzyme concentration in each well to 50 pM. The test compounds were prepared by serial dilution to be 20 times the final concentration in each well using DMSO as a solvent. Each concentration was then diluted 10 times in HEPES buffer before they were finally dispensed in each well. The substrate concentrations were prepared by diluting the stock solution in HEPES buffer.
The blank reactions were prepared by adding 50 µl of the highest concentration of the inhibitor in HEPES buffer to 25 µl of HEPES buffer. The control reaction on the other hand was prepared by adding 50 µl of 10% DMSO in HEPES buffer to 25 µl of the enzyme. The test compound reactions were prepared by adding 50 µl of the test compound in HEPES buffer to 25 µl of the enzyme. After 10 minutes incubation time, 25 µl of the peptide substrate was added to each of these groups to bring the final volumes to 100 µl. The fluorescence of the plate was recorded at λex of 360 nm and λem of 460 nm. The fluorescence readings were taken every 30 seconds for 10 minutes at 37°C, and the initial velocity of each reaction was calculated from the slopes as RFU/min.
The % Activity of the enzyme was calculated from the following equation:
% Activity = (VDrug/VControl×100)
The authors are thankful to MCPHS University for funding this research, no grant money was used.
- Gorrell MD, Gysbers V, McCaughan W (2001) CD26: A Multifunctional Integral Membrane and Secreted Protein of Activated Lymphocytes. Scand J Immunol 54: 249-264. [Crossref]
- Liu Y, Hu Y, Lu T (2012) Recent Advances in Non-Peptidomimetic Dipeptidyl Peptidase 4 Inhibitors: Medicinal Chemistry and Preclinical Aspects. Current Med Chem 19: 3982-3999. [Crossref]
- Nabeno M, Akahoshi F, Kishida H, Miyaguchi I, Tanaka Y, et al. (2013) A Comparative Study of the Binding Modes of Recently Launched Dipeptidyl Peptidase IV Inhibitors in the Active Site. Biochem Biophys Res Commun 434: 191-196. [Crossref]
- https://pdb101.rcsb.org/global-health/diabetes-mellitus/drugs/dpp4-inhibitor/dpp4
- Dooseop K, Kowalchick JE, Brockunier LL, Parmee ER, Eiermann GJ, et al. (2008) Discovery of Potent and Selective Dipeptidyl Peptidase IV Inhibitors Derived from β-Aminoamides Bearing Subsituted Triazolopiperazines. J Med Chem 51: 589-602. [Crossref]
- Brockunier L, He J, Colwell FLJr, Habulihaz B, He H, et al. (2004) Substituted Piperazines as Novel Dipeptidyl Peptidase IV Inhibitors. Bioorganic Med Chem Letter 14: 4763-4766. [Crossref]
- Chiara C, Tamas AM, Oliver R, Lukasz B (2014) Peptides Containing β-Amino Acid Patterns: Challenges and Successes in Medicinal Chemistry. J Med Chem 57: 9718-9739. [Crossref]
- Dallakyan S, Olson AJ (2015) Small-Molecule Library Screening by Docking with PyRx. Methods Mol Biol 1263: 243-250.
- Trott O, Olson AJ (2009) Auto Dock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J Comput Chem 31: 455-461. [Crossref]
- https://chemaxon.com/products/marvin
- O’Boyle NM, Banc M, James CA, Morley C, Vandermeersch T, et al. (2011) Open Babel: An Open Chemical Toolbox. J Cheminform 3: W504-W509. [Crossref]
- https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/visualization/
- George RL, Leiting B, Roy SR, Eiermann GJ, Beconi MG, et al. (2005) Dipeptidyl Peptidase IV Inhibition for the Treatment of Type 2 Diabetes: Potential Importance of Selectivity Over Dipeptidyl Peptidases 8 and 9. Diabetes 54: 2988-2994. [Crossref]