The fusion of autoantigens to the cholera toxin B subunit (CTB) adjuvant is a unique immunotherapeutic strategy known to protect experimental animals against autoimmune diseases. To understand the underlying mechanism of protection in humans, we examined the responses of human naïve T cells to co-culture with dendritic cells (DCs) treated with CTB fused to type 1 diabetes (T1D) autoantigens proinsulin or glutamic acid decarboxylase (CTB-INS and CTB-GAD). Our data show that CTB-INS and CTB-GAD autoantigen treated DCs inhibit the activation and proliferation of CD4+ T lymphocytes and weakly stimulate the activation and proliferation of CD8+ T cells. Naïve T cells co-cultured with CTB-INS treated DCs differentiated into anti-inflammatory CD4+ Th2 lymphocytes and CD4+, CD25+, CD-69+, FoxP3+, T regulatory cells (Tregs). Further, CTB-INS and CTB-GAD treated DCs stimulated T cell synthesis of the anti-inflammatory cytokines IL-10 and TGF-β while inhibiting T cell synthesis of pro-inflammatory cytokines IFN-g, and IL-17. Together, our data suggest CTB-INS and CTB-GAD induce a state of functional DC tolerance that can down regulate CD4+ pro-inflammatory Th cells and stimulate proliferation of Th and Treg cells that synthesize IL-10 and TGF-β anti-inflammatory cytokines. These factors may combine to establish an immune cell micro-environment that can prevent T1D.
cholera toxin B subunit, insulin, GAD, adjuvant, autoimmunity, dendritic cells, type 1 diabetes
Type 1 Diabetes mellitus (T1D) is an organ-specific autoimmune disease generally initiated in childhood but that may also occur later in life. The onset of T1D autoimmunity results in a progressive loss of insulin, resulting in a life-long insulin deficiency [1,2]. Acute and chronic increases in blood glucose are the clinical outcomes that lead to devastating complications. Blood vessel damage from the persistence of high blood glucose levels causes an increased risk for secondary circulatory and neural physiological complications that ultimately lead to amputation, blindness, kidney failure and an increased risk for heart attack, and stroke that result in a 15-20 yr decrease in expected life span [3]. Initial attempts to use oral insulin or the islet antigen GAD as an immune suppressive treatment offered a slight suppression of T1D autoimmunity, but the results were not consistent or sustained [4,5]. Boosting autoantigen inhibition of insulitis and hyperglycemia with adjuvants, showed that inhibition of T1D could be greatly enhanced by conjugation of islet autoantigens to the non-toxic B subunit of Vibrio cholerae enterotoxin (CTB) acting as the adjuvant [6-9]. Further experiments showed that CTB also acts as an efficient trans-mucosal carrier molecule for autoantigen induction of peripheral tolerance [10,11]. Toxin B subunits from a variety of bacterial and plant species when fused with pancreatic autoantigens could provide similar levels of autoantigen protection (50-65%) against the onset of T1D [12]. However, the molecular pathways responsible for generating the therapeutic effects of these fusion protein are largely unknown. The lack of adverse effects, in animals suggests their safety for application as interventions in humans.
Dendritic cells (DCs) considered to be the most potent of professional antigen presenting cells (APC) have been implicated in mediating the islet β-cell destruction observed in T1D. Through antigen presentation to cognate T cells, DCs may occupy an important position in the onset and regulation of innate and adaptive immune responses leading to T1D through their unique ability to prime naïve T helper cells for differentiation into effector T cells [13]. Regulation of these pro-inflammatory and anti-inflammatory processes is pivotal for maintenance of immunological homeostasis and in particular, the control or suppression of potentially autoimmune processes directed toward islets [14-17]. In the non-obese diabetic (NOD) mouse model of T1D, DCs presenting β-cell autoantigens migrate to the pancreas where they initiate inflammatory autoreactive effector Th1 cell responses that cause autoreactive B cells, CTLs and macrophages to migrate to and infiltrate the islets where they secrete autoreactive antibodies, inflammatory oxidative compounds NO, O2, H2O2, perforins and proteases in addition to several pro-inflammatory cytokines (IL-1 β, TNF-ᾳ, TNF-β) [5-7]. These toxic immune cell responses combine to trigger the onset of chronic pancreatic inflammation (insulitis), which ultimately destroys the β-cells reducing the levels of insulin required to meet the growing body’s metabolic requirements for energy and ultimately resulting in diabetes onset [18]. Recently, we showed that human dendritic cell activation was blocked by diabetes autoantigens linked to the cholera toxin B subunit. [19]. However, the mechanism responsible for this immune response remains unclear.
Construction and isolation of CTB-INS and CTB-GAD35 fusion proteins from E. coli
DNA fragments encoding 105 bp (amino acids 509-543), proximal to the carboxyl-terminus of the glutamic acid decarboxylase (GAD65 kDa) gene, designated GAD35 (35 aa) and 258 bp encoding the human proinsulin (INS) gene, were genetically linked to the carboxyl-terminus (309 bp) of the cholera toxin B subunit (CTB), to generate the fusion gene fragments CTB-GAD35 and CTB-INS respectively (Figure 1A-1C). The CTB–autoantigen fusion genes were cloned into E. coli expression vector pRSET (A) (Invitrogen™, Carlsbad, CA), under control of the bacteriophage T7 promoter in order to achieve high levels of transgene expression (Figure 1B). The pRSET vector also contains an oligonucleotide encoding a series of 6 histidines immediately upstream of CTB permitting Nickel column isolation of the transgene product based on the affinity of histidine for a nickel substrate fixed on the column. Selective transformed bacterial clones were assessed by DNA sequence analysis to confirm the in-frame linkage of CTB, CTB-INS and CTB-GAD35 DNA fragments the recombinant expression vectors were introduced into the E. coli producer strain BL21 (DE3)pLysS (Invitrogen, Carlsbad, CA) by electroporation as described earlier [12,19].
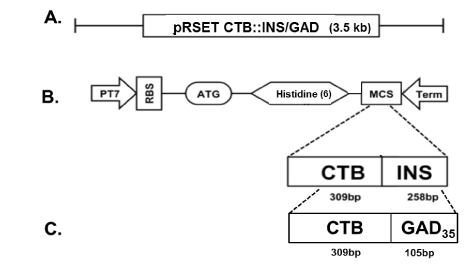
Figure 1.E coli expression vectors containing the CTB-INS and CTB-GAD fusion genes.
In Panel (A): Designation of the E. coli expression vectors in which the vector pRSET A contains the CTB-INS or CTB-GAD35 fusion genes. Panel (B): Map of the E. coli expression vector pRSET containing a cDNA fragment of the entire cholera toxin B subunit gene (CTB), linked in a c-terminal fusion with the complete human proinsulin gene (INS) and inserted into the plasmid multiple cloning site (MCS), to form the gene fusion CTB-INS (567bp). Panel (C): Plasmid map of E. coli expression vector pRSET-CTB-GAD containing a cDNA fragment encoding the cholera toxin B subunit protein (CTB) linked in a C-terminal fusion with a 105 bp, DNA fragment encoding a 35 amino acid c-terminal peptide from the second most prominent pancreatic islet protein autoantigen glutamic acid decarboxylase (GAD). Recombinant fusion protein synthesis from the bacterial expression vectors pRSET-CTB-INS and pRSET-CTB-GAD are under control of the bacteriophage T7 promoter containing an oligonucleotide region encoding 6 histidine amino acid residues located immediately 5’ upstream of the CTB DNA sequence to allow nickel affinity column isolation of the recombinant fusion proteins. The recombinant plasmids were transformed into the E. coli recipient strain BL21 (DE3) pLysS, for maximum fusion protein expression and for histidine mediated isolation and purification of the recombinant proteins via a Maxwell 16 TM protein isolation robot (Promega Inc, Madison, WI, USA).
The recombinant CTB-INS and CTB-GAD35 fusion proteins expressed in E. coli were purified from bacterial homogenates by isolating the 6xHis-tagged fusion protein from lysed cells using nickel chelation column chromatography. Briefly, the transformed E. coli strain BL-21, was grown in 250 ml Luria Broth (LB) medium containing ampicillin (100 mg/ml) with shaking at 37oC. While still in log phase of growth, protein synthesis was stimulated by addition of 90 mg isopropyl β-D-1thiogalacto-pyranoside (IPTG), (Sigma Chemical Co. St. Louis, MO) to the bacterial culture. After 6 hr of continued incubation at 370C, the bacterial culture was pelleted by centrifugation in a SA-600 rotor for 10 min, at 5,000 rpm and 4oC, in a Sorvall RC5B centrifuge. The cell pellet was resuspended in 1.0 ml / tube of 10 mM HEPES buffer (pH 7.5), containing 100 mM imidazole. The cells were disrupted by sonication (3 x 10 sec bursts), at 10 W, with a Sonic 60 Dismembrator (Fisher Sci. Sunnyvale, CA). The recombinant proteins were isolated and purified from the bacterial homogenate using a Maxwell Model 16 robotic protein purification system (Promega Inc.TM), according to the protein isolation protocol provided by the manufacturer (Promega Inc., Madison, WI). In order to obtain a pure protein product, the robot uses electromagnetically charged Magne-His Nickel-Iron alloy particles with an affinity for the 6-HIS tag linked to the N terminus of the recombinant fusion proteins. Imidazole was removed from the protein mixture by dialysis of the preparation against 2 x 1.0 Liter, 10 mM HEPES buffer (pH 7.5), for 4 - 8 hr at 4oC. The purity of the isolated CTB-INS (~23.4kDa) and CTB-GAD35 protein (~22kDa) was determined by analysis of protein electrophoretic mobility in a 12% polyacrylamide gel in comparison with protein molecular weight standards (BioRad Inc. Irvine, CA). The purified proteins were identified and confirmed by immunoblot analysis and stored at -20oC until used.
Human monocyte isolation and differentiation into dendritic cells
Monocytes were prepared from freshly collected human umbilical cord blood obtained from normal healthy placental donors following cesarean section in agreement with an established LLU IRB approved protocol. Briefly, umbilical cord blood (30 ml) was depleted of red blood cells, by incubation for 15 min with an ammonium chloride RBC lysis buffer (0.15 M NH4Cl, 1 mM KHCO3, and 0.1 mM Na2EDTA), centrifuged at low speed and the supernatant removed [19]. Erythrocyte depletion was repeated once to yield a tan leukocyte suspension. The CD14+ monocytes were isolated from the leukocyte mixture using a magnetic CD14 micro bead column as described by the manufacturer (Miltenyi Biotech, CA; catalog #130-050-201). Briefly, CD14+ monocyte was isolated by incubation of the leukocytes with anti-CD14 antibodies bound to magnetic beads in solution for 15 min at 4°C in the dark, followed by separation of the CD14+ cells from all other leukocytes by binding the beads to a magnetic LS column (Miltenyi Biotech) in a magnet (MidiMacs separator, Miltenyi Biotech, Auburn, CA). The isolated CD14+ monocytes were cultured in 6 cm well, non-pyrogenic polystyrene culture plates at (5 x 105 cells / ml / well) in RPMI 1640 culture medium (Mediatech Inc. Manassas, VA, USA), supplemented with 10% fetal bovine serum (FBS), 1 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 ng/ml human recombinant GM-CSF, (ProSpec-Tany TechnoGene, Rehovot Science Park, Israel) and 10 ng/ml human recombinant IL-4 (Preprotech, Rocky Hill, NJ), for 6 days at 37 °C in a humidified atmosphere of 5% CO2. The monocyte cell culture was fed at 2-day intervals by gentle replacement of 50% of the medium with fresh medium. The cell cultures were kept in their original wells until harvested to avoid induction of DC maturation caused by excessive mechanical stress associated with cell re-plating. After 6 days incubation, the percentage of monocyte-derived immature DCs (iDC) was determined by flow cytometry after surface marker staining with iDC-specific fluorescent Ab markers (CD14−HLA-DR+CD11c+).
Treatment of dendritic cells with CTB-INS and CTB-GAD fusion proteins
After 6 days incubation in culture medium supplemented with GMCSF and IL-4, the iDCs are treated with CTB (10µg/ml medium), GAD35 (10µg/ml), CTB-GAD35 fusion protein (20µg/ml), proinsulin (10µg/ml), CTB-INS (20µg/ml), or Phorbol Myristate Acetate and Ionomycin (PMA + Ionomycin) at (10 ng/ml RPMI medium), or alternatively as a positive control for activation, E. coli lipopolysaccharide (LPS) at (10 µg/ml RPMI medium). The CTB-autoantigen treated DCs were incubated for 48 hrs at 370C and 5% CO2. Following incubation, the DCs were stained for expression of DC surface markers (CD14- HLA-DR+ CD11c+), and the DC population was identified by flow cytometry. The DCs were then stained with antibodies against the DC activation markers, costimulatory factors CD86, CD80, CD40 and maturation factor CD83, as indicators of the state of DC activation (BD / Pharmingen, San Jose, CA USA). The cells were gated and analyzed for DC surface expression of co-stimulatory and maturation factor proteins by flow cytometry (MACSQuant, Miltenyi Biotec., Auburn, CA).
Isolation of human T cells from umbilical cord blood
Human umbilical cord blood (50 ml), was drawn from consenting normal healthy placental donors in compliance with LLU approved IRB protocols. White blood cells (WBCs) were separated from red blood cells (RBC) and platelet fractions with RBC lysis buffer as described above and the WBCs were pelleted at (2,000 RPM), for 10 min at 4oC, in a Beckman Coulter Allegra X-15R centrifuge, equipped with an SX4750 rotor. The CD3+ T cell fraction was positively selected from the WBCs using the MACS Pan T Cell Isolation Kit II protocol for indirect magnetic isolation of naïve T cells from human blood, as described by the manufacturer (Miltenyi Biotec. Auburn, CA). The purified T lymphocyte fraction was demonstrated by flow cytometric analysis to be > 99% pure. The T cells were re-suspended at 1 x 106 cells/ml in RPMI 1640 culture medium (Mediatech Inc. Manassas, VA, USA), supplemented with 10% FBS, 1 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. For long term storage, the purified T cells were adjusted to 10% DMSO and frozen at -800C until used in DC – T cell co-culture experiments.
Co-culture of T cells with CTB-INS and CTB-GAD treated dendritic cells
The monocyte derived iDCs were treated with CTB, INS, GAD, CTB-INS and CTB-GAD35 fusion protein and incubated for 48 hr at 370C to induce a DC response. The treated DCs were washed twice with PBS pelleted and mixed with thawed allogeneic T cells pre-activated for 48 hrs with anti-CD3 and anti-CD28 monoclonal antibodies prior to addition of the DCs. The DC:T cell ratio was 1:10, DCs (1x104 DC’s/well) + naïve T cells (1x105 T cells /well). The DCs - T cell mixture was suspended in 200ul of RPMI 1640 medium supplemented with 10% FBS, 1% L-glutamine, and 1% penicillin/streptomycin (Sigma-Aldrich; complete medium) /well in a 96-well microtiter plate and incubated at 370C with no further medium changes. T cell activation was determined 48 - 72 hrs after DC - T cell co-culture by harvesting the cell mixture from 2 wells / treatment into a 2.0 ml polypropylene culture tube. The T cells were stained for the appropriate surface markers and the T cell populations identified by flow cytometry.
T cell activation following incubation with CTB-INS and CTB-GAD treated DCs
The T cells were collected by low speed centrifugation and stained with fluorescent labeled anti-CD3, -CD4, -CD8, -CD25, and CD69, (a human transmembrane C-Type lectin protein that serves as an early T cells activation marker). The number of activated CD4+, CD8+ and regulatory T cell (Treg) cells are determined by flow cytometric identification of the following phenotypes: CD4+ or CD8+,CD25+, and CD127+ (Treg surface marker). In addition to staining for CD127, intracellular staining for the transcriptional activator Foxp3 was performed to identify native Treg cell populations. A portion of each DC-T cell co-culture was incubated for an additional 2 weeks with replacement of 10% of the medium every 2 days to allow amplification of the Treg cell population, prior to assessment of Treg populations by flow cytometry [20].
T cell proliferation following co-culture with DCs treated with CTB-INS and CTB-GAD
T cell subset proliferation was determined after 48 - 72 hr co-culture of naïve T cells with CTB-INS, CTB-GAD35 treated DCs as indicated above. The DC-T cell co-cultures were subjected to intracellular staining with fluorescent antibodies against the Kiel 67Kd protein (Ki-67), (a nuclear protein indicator of rRNA transcription indicative of cell proliferation). To confirm T cell proliferation during DC-T cell co-cultivation, a portion of each sample was treated with the fluorescent dye carboxyfluorescein diacetate, succinimidyl ester (CFSE), at an initial dye concentration of 3mM CFSE. After a 1 hr dye pulse, the T cells were washed with PBS to remove excess CFSE, returned to complete culture medium and incubated for an additional 6 days at 370C prior to flow cytometric assessment of dye fluorescence reduction in the cells. Each stepwise reduction in dye content in the cell curve was indicative of one synchronized cell division in the entire T cell population.
Cytokine biosynthesis in T cells co-cultured with CTB-INS and CTB-GAD treated DCs
For cytokine determinations, a total of 1x106 co-cultured T cells /ml were used for each cell proliferation and intracellular cytokine assay conducted in triplicate. After the 40 hr incubation with the DCs, the DC: T cell co-cultures were incubated for an additional 8 hrs in the presence of 3mM Monensin to sequester cytokine molecules in the T cell prior to analysis of cytokine synthesis. Intracellular cytokine staining was performed according to the manufacturer’s instructions (Caltag, CA). The T cell cultures were washed, and surface stained with fluorescent labeled anti-CD3 antibodies. Following staining for T cell class identification, the T cells were fixed in 0.5% paraformaldehyde in PBS, and permeabilized by the addition of 0.7% Tween-20 in PBS, to a final concentration of 0.14% and stained with primary Abs for the intracellular cytokines IFNγ, TGF-b, IL-17, FOXP3, IL-2 and IL-10. After washing (2x) with PBS, the cell cultures were fixed by resuspension in 1% paraformaldehyde and analyzed for the presence of intracellular cytokines using a MACSQuant flow cytometer (Miltenyi Biotec. Auburn, CA) present in the Center Core Facility.
T regulatory cell proliferation in CTB-INS treated DC - naïve T cell co-cultures
Because Treg cell differentiation and proliferation is often slower than observed for other T cell subsets, an experimental protocol was adopted to allow detection of small Treg cell numbers [20,21]. The Tregs were identified based on priming immature DCs with specific antigens (insulin) that stimulate cognate Treg cell development. Insulin specific Tregs were identified by measurement of down-regulation of CD3/TCR complexes on the T cell surface following a boost with the specific antigen. Thus, the protocol was optimized to allow detection of relatively infrequent Tregs specific for T1D autoantigens. Briefly, human peripheral blood mononuclear cells (PBMC) were separated from RBCs by RBC lysis. The lymphocytes, iDCs and naïve T cells were re-suspended at a concentration of 2.5 - 3.5 x 107 PBMCs / ml. Aliquots (200 µl), of the PBMC suspension were dispensed into triplicate wells of a 24-well plate and incubated with CTB-INS fusion protein (1 x 10-5 M final CTB-INS concentration), for 1 hour at 370C and 5% CO2. The cultures were diluted 10-fold (2.0 ml) with RPMI complete medium containing IL-2 (300 U/ml) and incubated at 370C for 14-28 days for Treg amplification. During this period, the cultures were fed at 2-3-day intervals using complete RPMI medium supplemented with IL-2 for optimal expansion of antigen-specific Tregs and split as required, taking care to maintain a high cell density (1.5-2.5 x 106 cells/ml. Following the 2-3 wk extended culture, antigen-specific Tregs were detected by incubating a maximum of 1x106 cells at 370C in 100 µl of fresh medium containing proinsulin at 1x10-4 M for 16 hr. The insulin boosted cells were then stained for T cell markers CD3+, CD4+, and Treg activation markers CD25+, CD127+ and FoxP3+ and analyzed by flow cytometry by gating on viable CD4+, CD3low T cells. T cell proliferation was assessed by flow cytometric detection of intracellular Ki-67. The DC-T co-cultured cell samples were harvested as described above and surface stained for T cell markers CD3+, CD4+, CD25+ and CD127+. [20]. To identify native Tregs, intracellular staining for the transcriptional activator Foxp3+ in place of CD127 was also carried out.
Statistical analysis
All cell samples were analyzed in triplicate. Statistical analyses of the experimental treatment groups was performed by measurement of the standard deviation and standard error of the mean in all assays used in this study and by application of Student’s T test or analysis of variance and (ANOVA) for comparison of 2 or more than 2 experimental functions or groups. T cell fluorescence intensity values and population percentages are presented as the mean ± 95% confidence interval. A value of p< 0.05 was considered to be statistically significant.
T cell activation is inhibited by CTB-autoantigen treated DCs
DC mediated T cell differentiation into autoreactive CD4+ or CD8+ cells plays an important role in the immune destruction of pancreatic β-cells. Populations of CD4+ and CD8+ activated T cells were detected in DC-T cell co-cultures by antibodies prepared against CD4 and CD8 surface markers (Figure 2A). Reduced levels of T cell activation marker CD69 were detected in CD4+ T cells but not in CD8+ T cells co-cultured with CTB-INS and CTB-GAD35 treated DCs (Figure 2B & 2C). In comparison with the negative control, (untreated DCs + T cells), iDCs treated with CTB-INS or CTB-GAD prior to T cell co-culture inhibited CD4+ and CD8+ T cell activation, more than each autoantigen or CTB alone (Figure 2D).
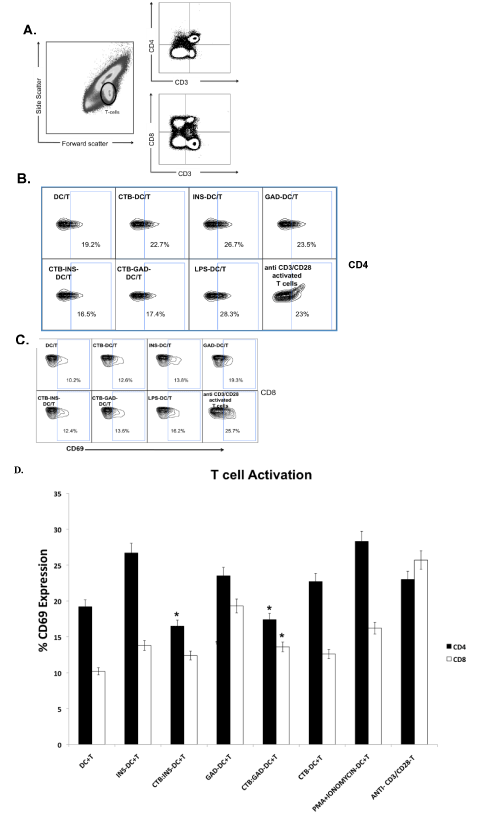
Figure 2.Inhibition of T cell activation by CTB-INS and CTB-GAD35 treated DCs.
Allogeneic naive T cells were activated by anti-CD3 and anti-CD28 antibodies (Ab) and co-cultured with iDCs previously treated with CTB-INS or CTB-GAD35. After incubation for 48 to 72 hr, the number of activated CD4+ and CD8+ T cells was determined by flow cytometric detection of the T lymphocyte activation marker CD69. Panel (A): is a representative flow cytometry plot of CD4+ and CD8+ T cell populations following co-cultivation with CTB-INS and CTB-GAD. T Lymphocytes were gated based on forward and side scatter (left panel) and by increased expression of CD3+ and CD4+ (top right), or CD8+ (bottom right). Panel (B): Is a representative FACS plot showing CD69 expression after gating on CD4+ T cells. Panel (C): is a FACS plot of CD69 expression after gating on CD8+ T cells. Panel (D): is a bar graph representing the percent of CD69+ activated CD4+ and CD8+ T cells detected following incubation of naïve T cells with DCs treated with INS, GAD, CTB-INS or CTB-GAD fusion proteins. The data represent the means and SEM for 4 independent experiments for each protein. Asterisks indicate a statistically significant reduction in T cell synthesis of CD69 marker protein (p<0.05) for CTB-autoantigen fusion protein samples in comparison with autoantigen alone samples.
T cell proliferation is reduced by CTB-INS and CTB-GAD35 treated DCs
T cell differentiation and proliferation were examined following naïve T cell co-cultivation with iDCs treated with CTB, INS, GAD, CTB-INS, CTB-GAD35 or PMA + Ionomycin. Increases in CD4+ and CD8+ T cell numbers were measured by flow cytometry after labeling the T cells with fluorescent Abs against the cell proliferation marker protein KI-67 (Figure 3A & 3B). Naïve T cells co-cultivated with iDCs treated with CTB or PMA + Ionomycin stimulated maximum proliferation of both CD4+ and CD8+ T cells. Naive T cells co-cultured with CTB-INS or CTB-GAD35 treated DCs showed reduced levels of KI-67 in comparison with T cells cultivated with DCs treated with INS or GAD. This experimental result indicated that CTB-INS and CTB-GAD treated DCs reduce CD4+ and CD8+ T cell proliferation (Figure 3A & 3B). In comparison with INS stimulation of T cell proliferation, CTB-INS treatment of iDCs reduced CD4+ T cell proliferation about 10% and CD8+ T cell proliferation by about 2%. In comparison, CTB-GAD treated DCs reduced CD4+ proliferation by about 7% and was similar to CTB-INS, reducing CD8+ T cell proliferation by about 2%. To confirm this result, T cell proliferation was measured by incubation of DC – T cell co-cultures with the fluorescent dye CFSE (Figure 3C). Each T cell division is represented by arrows identifying the presence of small plateaus of decreasing CFSE fluorescence during co-cultivation of naïve T cells with DCs treated with the fusion proteins, INS, GAD and PMA + Ionomycin. As observed with KI-67 staining of the T cells, reductions detected in CFSE staining confirm that both CTB-INS and CTB-GAD inhibit CD4+ and CD8+ T cell proliferation.
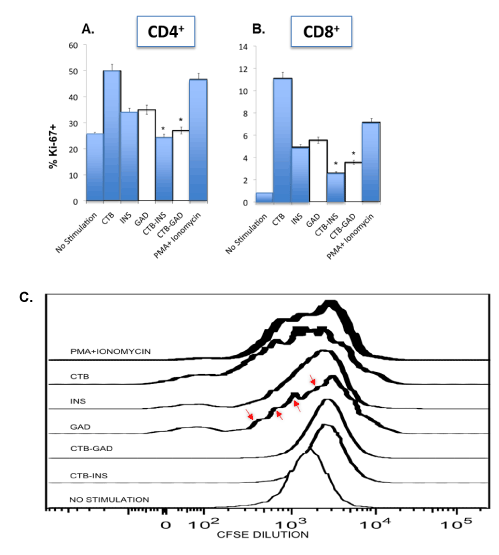
Figure 3.CTB-INS and CTB-GAD treated DCs inhibit T cell proliferation.
INS, GAD, CTB-INS, CTB-GAD and PMA-Ionomycin treated iDCs were co-cultured with naive allogeneic T cells for 4 days in RPMI culture medium. After harvesting the T cells by centrifugation, they were stained with CD3+, CD4+ and CD8+ surface markers and for intracellular markers to detect Tregs and with Ki-67 to detect T cell proliferation, prior to flow cytometry. T cells (CD3+) were gated into CD4+ and CD8+ subsets and the percentage of Ki-67+ cells determined. In Panel (A), the level of T cell proliferation was plotted for CD4+ T helper cells and in Panel (B), the proliferation of CD8+ cytotoxic T cells (CTLs). The bars in each graph represent the mean and standard error of the mean (SEM) taken from three separate DC-T cell co-culture experiments. Asterisks (*) over the bars represent detection of a significant difference (p<0.05), in the expression of Ki-67+ in T cells incubated with DCs inoculated with CTB-autoantigen fusion proteins in comparison with T cells incubated with DCs inoculated with INS or GAD autoantigens. In a complementary experiment, naïve T cell cultures were labeled for 1 hr with the fluorescent dye CFSE at day 4 of incubation with CTB, INS, GAD, CTB-INS, CTB-GAD and PMA + Ionomycin and treated DCs Panel (C). After staining and washing the T cells to remove excess CFSE dye, the cells were incubated in culture medium without CFSE for an additional 6 days. The cultures were analyzed for dye reduction by flow cytometry to determine the approximate number of T cell divisions identified by the periodic reduction in cellular CFSE fluorescence intensity (arrows indicating small plateaus).
CTB-INS treated DCs induce regulatory T cell proliferation
Because regulatory T cells (Tregs) can mediate immunological tolerance via inhibiting DC generated pro-inflammatory responses [22], we assessed whether CTB-INS or CTB-GAD could induce DC tolerance by stimulating the proliferation of Tregs. Naïve allogeneic T cells were co-cultivated with CTB-INS treated DCs for 2-3 wk. the T cells were treated with an insulin boost to amplify insulin specific T cells. After an additional week of culture, flow cytometric analysis of viable CD4+, CD25+, FoxP3+, CD3 low, autoantigen specific T cells revealed a modest 2.1% increase in Tregs (Figure 4A-4C). In comparison with T cells co-cultured with INS or CTB treated DCs, dendritic cells treated with CTB-INS showed an increase in Treg proliferation (KI-67 staining). An approximate 4-fold increase in percent Tregs expressing Ki-67 was detected in CTB-INS vs INS stimulated DCs co-cultivated with naïve T cells (Figure 4D), Triplicate T cell samples were tested with a probability value of (p<0.05).
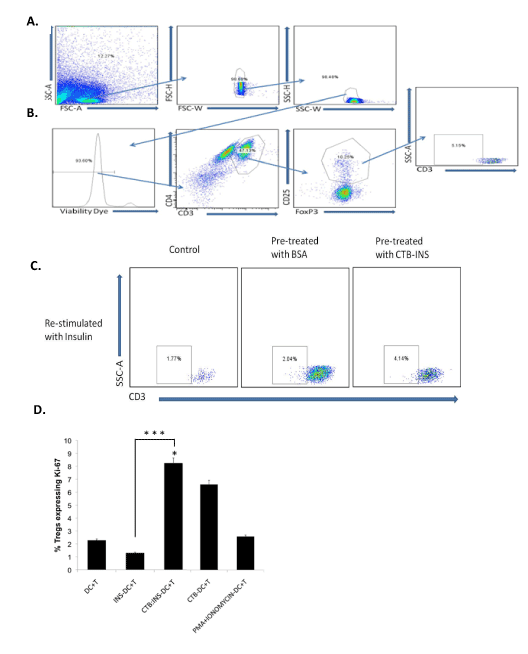
Figure 4.CTB-insulin stimulates Treg cell proliferation.
PBMC lymphocytes from human umbilical cord blood were pulsed with CTB-INS fusion protein and incubated in RPMI medium with human serum and IL2 for 3 wk to amplify antigen specific Treg cells. Following a proinsulin boost 2-3 wk after the CTB-INS pulse, the T cells were assessed for viability by flow cytometry forward and side scatter analysis (SSC-A), Panel (A): top three frames. Cell viability was assessed by T cell staining with eFluor450, first frame from left in Panel (B). The cells were stained and gated for surface expression of CD4+ and CD3+ Panel (B): second frame from left. The CD4+, CD3 high cells were selected and the CD25 high FoxP3 positive Tregs gated upon Panel (B): 3rd frame from the left. The CD3 low cell population (putative insulin specific Tregs), were gated on in the 4th frame from left, Panel (B). A comparison was made between lymphocytes that did not receive the vaccine primer dose but were later “boosted” with insulin Panel (C): left frame), and T cells that were primed with BSA and boosted with insulin Panel (C): center frame and finally, lymphocytes primed with CTB-INS fusion protein and boosted with proinsulin Panel (C): right frame. In Panel (D): the proliferation of Tregs was described graphically following flow cytometric determination of the percentage of activated Tregs expressing Ki-67. Differences observed in the proliferation of Treg populations were determined by ANOVA (p<0.05). The bar graph represents the percentage of Treg cells expressing the Ki-67 transcription factor as an indicator of cell division. Individual columns represent the means and SEM of three independent experiments. The asterisks (*) represent significant differences in the expression of Ki-67 (cell proliferation) in Tregs cells for T cells incubated with CTB-autoantigen fusion protein inoculated DCs in comparison with T cells incubated with DCs inoculated with the single autoantigens alone (p<0.05).
CTB-autoantigen treated DCs stimulate T cell cytokine synthesis
Both CTB-INS and CTB-GAD35 treated iDCs clearly stimulated naïve CD3+ T cell biosynthesis of the anti-inflammatory cytokine IL-10. However, only CTB-GAD treated DCs stimulated synthesis of the major anti-inflammatory cytokine TGF-b (Figure 5). In comparison with DC + T cell control co-cultures, CTB-INS treated iDCs inhibited T cell biosynthesis of IFN-g, and IL-17 pro-inflammatory cytokines (p<0.05) (Figure 5). Naïve T cells co-cultured with DCs treated with CTB did not stimulate IL-10 or TGF-β synthesis, however, DCs treated with CTB-INS and CTB-GAD35 stimulated T cell biosynthesis of IL-10 and TGF-β, cytokines characteristic of Th2, Tr1 (IL-10) and Th3 (TGF-β) regulatory T cells. Thus, our experimental data indicate that in general, CTB-autoantigen treated DCs induce the differentiation of anti-inflammatory T cells. Further, our data suggest that CTB-INS and CTB-GAD treated DCs induce DC tolerance by increasing T cell secretion of IL-10 and TGF-β anti-inflammatory cytokines and the number of Tregs. The decrease in T cell secretion of pro-inflammatory cytokines IFN-λ and IL-17 detected in T cells co-cultured with CTB-INS or CTB-GAD help to confirm this hypothesis.
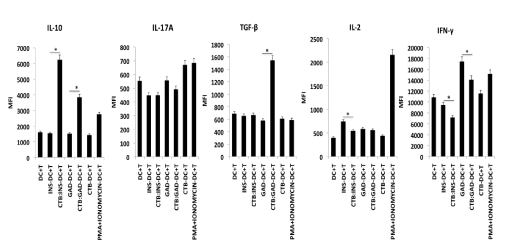
Figure 5.CTB-INS and CTB-GAD treated DC stimulation of T cell cytokine synthesis.
Naïve CD3+ T cells isolated from human umbilical cord blood were activated with anti-CD3/CD28 antibodies and co-cultured with iDCs previously treated with CTB, INS, GAD, CTB-INS, CTB-GAD35 or PMA + Ionomycin, The DC -T cell co-cultures were incubated at 370C for 48 hr and Monensin (0.3 μM), was added to each T culture at 40 hr for the final 8 hrs of incubation to permit accumulation of cytokines within the T cells prior to examination of their intracellular cytokine levels by flow cytometry. The bar graphs represent T cell biosynthesis of cytokines IL-10, TGF-b, IFN-g, IL-2 and IL-17 following incubation of the naïve T cells with DCs treated with the CTB-autoantigen fusion proteins indicated above. The Y axis represents the T cell mean fluorescence intensity levels (MFI) detected. The X axis indicates the DC-T cell co-culture combinations tested. Statistical significance among triplicate samples of each DC-T cell co-culture was calculated based on a value of p <0.05.
Earlier studies show that DCs induce a state of immune tolerance that quiets overactive immune responses that stimulate autoimmunity [23,24]. While both CTB-INS and CTB-GAD fusion proteins inhibit type 1 diabetes in NOD mice, this result remains to be confirmed in human T1D patients [25-27]. The rationale that DC tolerance down regulates inflammation suggests the activation of DCs could be responsible for stimulating T1D. Our data reinforces the results of earlier studies, by demonstrating CTB-INS and CTB-GAD treated DCs can induce a state of functional tolerance in DCs that promotes T cell differentiation into anti-inflammatory effector and regulatory T cell populations [28-30]. While CTB, treated DCs induce the proliferation of pro-inflammatory T cells, CTB-INS, treated DCs reduce the numbers of CD4+ and CD8+ T cells and stimulate the differentiation of Th2 cells and induce T cell synthesis of anti-inflammatory cytokines IL-10 and TGF-β. Because T cell fixation is required, FOXP3 cannot be easily used as a marker to identify native Tregs. [31-34]. To circumvent this problem, the IL-7 receptor a-chain CD127 was used in conjunction with CD4+ and CD25+ as markers to identify native Tregs [34]. Tregs inhibit DC induction of pro-inflammatory T cells responsible for autoimmunity [35,36]. CTB-autoAg treated DC stimulation of T cell IL-10 secretion suggests that Th2 and/ or Tr1 regulatory cells may down regulate T1D. The downregulation of IL-17 in T cells co-cultivated with CTB-INS and CTB-GAD treated DCs suggests CTB-autoantigen fusion proteins may inhibit T1D by blocking synthesis of the pro-inflammatory cytokine [37-39]. Down regulating IL-17 may have broad implications for the inhibition of autoimmunity, as IL-17 was found in the serum and tissues of both tissue specific and systemic autoimmune diseases including arthritis, lupus, and multiple sclerosis [38,40-42]. Our recent preliminary data (not shown), indicates that CTB-GAD35 inhibits DC secretion of IL-6, a pro-inflammatory cytokine responsible for maintaining the balance between Th17 cells and Tregs. Thus, CTB-autoAg inhibition of IL-6 may reduce the contribution of Th17 cells to T1D progression [43-46].
Together, our data show that treatment of human DCs with CTB-INS or CTB-GAD fusion proteins can protect against T1D autoimmunity by: 1) inhibiting the differentiation and proliferation of CD4+ and CD8+ effector T cells. 2) by stimulating the proliferation of regulatory T cells, 3) by inducing T cell synthesis of anti-inflammatory cytokines and, 4) by inhibiting T cell production of pro-inflammatory cytokines. The continued examination of CTB-autoantigen fusion protein efficacy for protection against T1D autoimmunity in vivo will help to enable this effective “off the shelf” molecular therapy to become a key strategy for the prevention and treatment of T1D.
We would like to thank the Loma Linda University Medical Center, Division of Labor and Delivery, for providing the umbilical cord blood needed for our experiments and Dr. Kimberly Payne for constructive criticisms for improvement of the manuscript. This project was supported in part by funding from grant 1-2000-812 from the Juvenile Diabetes Foundation to Dr. William Langridge and an R21 grant DK-99-013 awarded to William Langridge and Istvan Fodor from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) branch of the National Institutes of Health.
- Eisenbarth GS (1986) Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med 314: 1360-1368. [Crossref]
- Tisch R, McDevitt H (1996) Insulin-dependent diabetes mellitus. Cell 85: 291-297. [Crossref]
- Libby P, Nathan DM, Abraham K, Brunzell JD, Fradkin JE, et al. (2005) Report of the National Heart, Lung, and Blood Institute-National Institute of Diabetes and Digestive and Kidney Diseases Working Group on Cardiovascular Complications of Type 1 Diabetes Mellitus. Circulation 111: 3489-3493. [Crossref]
- Zhang J, Yuen BH, Currie WD, Leung PC (1991) Suppression of luteinizing hormone secretion by atrial and brain natriuretic peptides in ovariectomized rats. Endocrinology 129: 801-806. [Crossref]
- Czerkinsky C, Anjuere F, McGhee JR, George-Chandy A, Holmgren J, et al. (1999) Mucosal immunity and tolerance: relevance to vaccine development. Immunol Rev 170: 197-222. [Crossref]
- Arakawa T, Yu J, Chong DK, Hough J, Engen PC, et al. (1998) A plant-based cholera toxin B subunit-insulin fusion protein protects against the development of autoimmune diabetes. Nat Biotechnol 16: 934-938. [Crossref]
- Arakawa T, Chong DK, Langridge WH (1998) Efficacy of a food plant-based oral cholera toxin B subunit vaccine. Nat Biotechnol 16: 292-297. [Crossref]
- Arakawa T, Yu J, Langridge WH (1999) Food plant-delivered cholera toxin B subunit for vaccination and immunotolerization. Adv Exp Med Biol 464: 161-178. [Crossref]
- Bergerot I, Ploix C, Petersen J, Moulin V, Rask C, et al. (1997) A cholera toxoid-insulin conjugate as an oral vaccine against spontaneous autoimmune diabetes. Proc Natl Acad Sci U S A 94: 4610-4614. [Crossref]
- Shreedhar VK, Kelsall BL, Neutra MR (2003) Cholera toxin induces migration of dendritic cells from the subepithelial dome region to T- and B-cell areas of Peyer's patches. Infect Immun 71: 504-509. [Crossref]
- Sun JB, Holmgren J, Czerkinsky C (1994) Cholera toxin B subunit: an efficient transmucosal carrier-delivery system for induction of peripheral immunological tolerance. Proc Natl Acad Sci U S A 91: 10795-10799. [Crossref]
- Carter JE 3rd, Yu J, Choi NW, Hough J, Henderson D, et al. (2006) Bacterial and plant enterotoxin B subunit-autoantigen fusion proteins suppress diabetes insulitis. Mol Biotechnol 32: 1-15. [Crossref]
- Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392: 245-252. [Crossref]
- Pulendran B, Smith JL, Caspary G, Brasel K, Pettit D, et al. (1999) Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc Natl Acad Sci U S A 96: 1036-1041. [Crossref]
- Pulendran B, Kumar P, Cutler CW, Mohamadzadeh M, Van Dyke T, et al. (2001) Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J Immunol 167: 5067-5076. [Crossref]
- Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, et al. (2003) Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity 19: 47-57. [Crossref]
- Kapsenberg ML (2003) Dendritic-cell control of pathogen-driven T-cell polarization. Nat Rev Immunol 3: 984-993. [Crossref]
- Piccinni MP, Beloni L, Livi C, Maggi E, Scarselli G, et al. (1998) Defective production of both leukemia inhibitory factor and type 2 T-helper cytokines by decidual T cells in unexplained recurrent abortions. Nat Med 4: 1020-1024. [Crossref]
- Odumosu O, Payne K, Baez I, Jutzy J, Wall N, et al. (2010) Suppression of dendritic cell activation by diabetes autoantigens linked to the cholera toxin B subunit. Immunobiology 216: 447-456. [Crossref]
- Ebert LM, MacRaild SE, Davis ID, Cebon J, Chen W (2012) A novel method for detecting antigen-specific human regulatory T cells. J Immunol Methods 377: 56-61. [Crossref]
- Valitutti S, Lanzavecchia A (1997) Serial triggering of TCRs: a basis for the sensitivity and specificity of antigen recognition. Immunol Today 18: 299-304. [Crossref]
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M (1995) Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155: 1151-64. [Crossref]
- Steinman RM, Turley S, Mellman I, Inaba K (2000) The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med 191: 411-416. [Crossref]
- Wakkach A, Fournier N, Brun V, Breittmayer JP, Cottrez F, et al. (2003) Characterization of dendritic cells that induce tolerance and T regulatory 1 cell differentiation in vivo. Immunity18: 605-617. [Crossref]
- Gong Z, Long X, Pan L, Le Y, Liu Q, et al. (2009) Cloning, expression, purification and characterization of the cholera toxin B subunit and triple glutamic acid decarboxylase epitopes fusion protein in Escherichia coli. Protein Expr Purif 66: 191-197. [Crossref]
- Gong Z, Jin Y, Zhang Y (2007) Suppression of diabetes in non-obese diabetic (NOD) mice by oral administration of a cholera toxin B subunit-insulin B chain fusion protein vaccine produced in silkworm. Vaccine 25: 1444-1451. [Crossref]
- Denes B, Krausova V, Fodor N, Timiryasova T, Henderson D, et al. (2005) Protection of NOD mice from type 1 diabetes after oral inoculation with vaccinia viruses expressing adjuvanted islet autoantigens. J Immunother 28: 438-448. [Crossref]
- Battaglia M, Stabilini A, Draghici E, Migliavacca B, Gregori S, et al. (2006) Induction of tolerance in type 1 diabetes via both CD4+CD25+ T regulatory cells and T regulatory type 1 cells. Diabetes 55: 1571-1580. [Crossref]
- Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, et al. (2006) Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev 212: 28-50. [Crossref]
- Levings MK, Gregori S, Tresoldi E, Cazzaniga S, Bonini C, et al. (2005) Differentiation of Tr1 cells by immature dendritic cells requires IL-10 but not CD25+CD4+ Tr cells. Blood 105: 1162-1169. [Crossref]
- Khattri R, Cox T, Yasayko SA, Ramsdell F (2003) An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol 4: 337-342. [Crossref]
- Hori S, Nomura T, Sakaguchi S (2003) Control of regulatory T cell development by the transcription factor Foxp3. Science 299: 1057-1061. [Crossref]
- Fontenot JD, Gavin MA, Rudensky AY (2003) Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 4: 330-336. [Crossref]
- Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, et al. (2006) CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med 203: 1701-1711.
- Yudoh K, Matsuno H, Nakazawa F, Yonezawa T, Kimura T (2000) Reduced expression of the regulatory CD4+ T cell subset is related to Th1/Th2 balance and disease severity in rheumatoid arthritis. Arthritis Rheum 43: 617-627. [Crossref]
- Hall AM, Ward FJ, Vickers MA, Stott LM, Urbaniak SJ, et al. (2002) Interleukin-10-mediated regulatory T-cell responses to epitopes on a human red blood cell autoantigen. Blood 100: 4529-4536. [Crossref]
- Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, et al. (2003) Increased expression of interleukin 17 in inflammatory bowel disease. Gut 52: 65-70. [Crossref]
- Nakae S, Nambu A, Sudo K, Iwakura Y (2003) Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol 171: 6173-6177. [Crossref]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, et al. (2006) IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol 177: 566-573. [Crossref]
- Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, et al. (2008) Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 172: 146-155. [Crossref]
- Wong CK, Ho CY, Li EK, Lam CW (2000) Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2 cytokine (IL-4) concentrations in patients with systemic lupus erythematosus. Lupus 9: 589-593. [Crossref]
- Hofstetter HH, Ibrahim SM, Koczan D, Kruse N, Weishaupt A, et al. (2005) Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol 237: 123-130. [Crossref]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, et al. (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441: 235-238. [Crossref]
- Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, et al. (2006) Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441: 231-234. [Crossref]
- Yokota S, Imagawa T, Mori M, Miyamae T, Aihara Y, et al. (2008) Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet 371: 998-1006. [Crossref]
- Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, et al. (2008) Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 112: 3959-3964. [Crossref]