Breast cancer is a major cause of morbidity in women worldwide, thus necessitating the identification of novel therapeutic options. The present study aimed to investigate the inhibitory effects of a crude flavonoid extract (CFENS) isolated from the medicinal herb Nigella sativa on the proliferation of MCF- 7 human breast cancer cells and to elucidate the mechanism of action. The MCF-7 cell viability was examined by an MTT assay. Fluorescent microscopy, flow cytometry analyses, and agarose gel electrophoresis were carried out to assess the pro-apoptotic potentiality of CFENS. Western blot analyses were done to detect gene expression. The findings showed that CFENS dose-dependently inhibited proliferation and induced apoptosis in MCF-7 cells. Typical morphologic and biochemical changes of apoptosis, including cell shrinkage and detachment, nuclear condensation, and DNA damage, were observed after the CFENS treatments. CFENS triggered ROS accumulation, GSH depletion, disruption of mitochondrial membrane potential, activation of caspases-3/7 and -9, and an increase in the Bax/Bcl-2 ratio in MCF-7 cells. In addition, CFENS induced cell cycle arrest, upregulated the expression levels of p53 and p21 proteins, and downregulated the expression of cyclin D1. These findings indicate that CFENS may help prevent breast cancer and may potentially be a useful agent for the treatment of certain malignancies.
medicinal herbs, oxidative stress, mitochondrial potential, apoptosis
Despite the efforts of innumerable researchers to ameliorate the dismal outcomes of cancer, this disease continues to be a major public health problem and is the second leading cause of death worldwide [1]. In particular, breast cancer is one of the most common cancers endangering women and accounts for 30% of all newly diagnosed cancers [2]. Breast cancer refers to a heterogeneous group of diseases that are clinically subdivided into hormone estrogen receptor–positive, epidermal growth factor receptor 2–positive, and triple-negative breast cancers [3]. Among these subtypes, the estrogen receptor–positive phenotype is the most prevalent, comprising ~75% of all breast cancers, and has a more favorable prognosis and pattern of recurrence, with endocrine therapy being the pillar of treatment. Although the existing therapies for breast cancer allow a significant decrease in mortality, a large number of patients fail to respond to initial therapy (de novo resistance) or develop resistance after prolonged treatment (acquired resistance), thus limiting the usefulness of the drugs given [4]. Therefore, it is necessary to find novel therapeutic approaches for this cancer.
For the past decades, researchers have tried to elucidate the etiology of various cancer phenotypes by searching for the molecular target(s) of the disease, which could then be targeted by drugs. However, the elucidation of the molecular mechanisms underlying neoplastic transformation and progression has resulted in the understanding that breast cancer, as well as most cancer phenotypes, evolves due to the dysregulation of multiple genes and multiple cell signaling pathways [5]. On the other hand, most of the current chemotherapeutics, including approved molecular targeted therapy–based drugs, act as mono-target molecules. Therefore, the complex interactions and cross talk among various cancer-associated genes, proteins, and other molecules in a living body compromise the therapeutic effect of mono-target drugs, even after the compound has been screened and tested in the laboratory and in animals. In this regard, network models suggest that partial inhibition of a surprisingly small number of targets can be more efficient than complete inhibition of a single target [6]. Thus, there is a need to identify active compounds with the potential to target two or more pathways at once or a few players in the same pathway for the treatment of cancer. Therefore, researchers have been turning their attention to developing multi-target interventions by using combination therapies of active compounds. To this end, evidence-based herbal medicines could be a reservoir for developing multi-target interventions. Accumulated research reports have shown that unlike widely used pharmaceutical drugs, which act as mono-target molecules, crude extracts of herbal remedies have multi-target molecules that could synergistically target multiple pathways involved in the initiation and progression of cancer [7].
Among medicinal herbs, the seeds and oil of Nigella sativa, in the Ranunculaceae family, have attracted the interest of medical scientists. The seeds, commonly known as black seeds, black cumin, or black caraway seeds, have been used in traditional medicine as a remedy for a long list of ailments [8]. Importantly, the seeds and oil of the herb are characterized by a very low degree of toxicity [9]. Although the biological activity of the herb is essentially attributed to its main component in essential oil, known as thymoquinone [10], numerous active compounds have more recently been isolated and identified in N. sativa. For example, the herb has been found to contain a considerable amount of flavonoids [11,12]; nonetheless, no report thus far has elucidated the anticancer activity of these flavonoids.
Flavonoids are a group of bioactive compounds extensively found in foodstuffs of plant origin. They possess a remarkable spectrum of pharmacologic properties, such as antimicrobial, antiviral, anti-inflammatory, antiallergic, analgesic, antioxidant, and hepato-protective activities. The use of flavonoids for both cancer prevention and treatment has been well documented over the past decade [13,14]. Numerous epidemiologic studies have validated the inverse relation between consumption of flavonoids and risk of breast cancer, as well as other cancer phenotypes [15,16]. Flavonoids play a critical role in controlling cancer at multiple levels, including the inactivation of pro-carcinogens, inhibition of metastasis and angiogenesis, stimulation of immune response against cancer cells, sensitization of cancer cells to cytotoxic drugs, and control of the cell cycle [13,16]. Importantly, flavonoids have the potentiality to induce apoptosis in cancer cells through modulation of a number of key elements in signal transduction pathways linked to apoptosis, modulation of the Bcl-2 protein family expression, activation of caspases, and DNA fragmentation [17].
Apoptosis is a key regulator of physiologic growth control and regulation of tissue homeostasis. It is one of the most potent defenses against cancer because it eliminates potentially deleterious, mutated cells, and impairment of apoptotic pathways is a major hallmark of cancer growths [18]. Consistently, the successful eradication of cancer cells through induction of apoptosis is the ultimate aim of chemotherapy [19]. Apoptosis is generally accepted to be initiated by two interconnected signaling pathways: the death receptor (extrinsic) pathway, which operates through cell surface death receptors, and the mitochondrial (intrinsic) pathway. However, both pathways converge on a common effector: caspase-3 [20]. On activation of the death receptor, a series of signaling protein interactions occurs, which ultimately leads to activation of one or more members of the caspase family of cell death proteases, classically caspase-8 [20]. The intrinsic pathway is initiated by anticancer drugs, growth factor withdrawal, or hypoxia. Any of these stimuli induce permeabilization of the outer mitochondrial membrane, leading to the release of apoptogenic factors from the mitochondrial intermembrane space into the cytosol. However, the permeabilization of the mitochondrial membrane is governed by Bcl-2-family proteins [21]. This family consists of antiapoptotic proteins (e.g., Bcl-2), as well as a number of proapoptotic molecules (e.g., Bax). Overexpression of the Bcl-2 protein blocks mitochondrial outer membrane permeabilization and inhibits apoptosis, whereas increased expression of the Bax protein offsets the role of Bcl-2 [12]. The release of apoptogenic factors into the cytosol triggers the activation of an initiator caspase (caspase-9), which cleaves and activates downstream effector caspases, in particular caspase-3. Caspase-3 then cleaves a large number of intracellular substrates, leading to the emergence of the morphologic changes of apoptosis [22].
An increasing number of studies has shown that excessive reactive oxygen species (ROS) can directly induce permeabilization of the outer mitochondrial membrane, leading to the release of apoptogenic factors, propagation of the apoptotic cascade, and execution of cell death [23]. ROS are constantly generated and eliminated in all aerobic cells; they act as secondary messengers in cell signaling and are required for various biological processes in normal cells [24]. ROS normally exist in balance with biochemical antioxidants in all aerobic cells [25]. However, when this critical balance is disrupted by excessive ROS production and/or antioxidant depletion, oxidative stress may ensue [25]. Intracellular accumulation of ROS plays a central role in developing cancer [26]. However, ROS play a dual role in carcinogenesis because they can either propagate cancers or induce death of cancer cells through apoptosis. A fact that makes ROS an important target for developing antitumor drugs and many chemotherapeutics is that its induction has been found to selectively kill cancer cells [27]. To protect themselves against ROS damage, cells have a robust antioxidant system, including antioxidant enzymes and redox-regulating proteins. Reduced glutathione (GSH) is the major redox-regulating protein that maintains redox homeostasis and protects cells against ROS stress and apoptotic cell death [28]. Although GSH plays a role in cell survival, increased levels of cellular GSH may promote the survival of tumor cells and confer resistance to cancer chemotherapeutic drugs [29]. Therefore, targeting cellular GSH in cancer cells may increase the cell sensitivity to cytotoxic agents [30].
The success stories of using crude extracts as chemopreventive agents and the fact that these extracts hold multi-target constituents have led us to investigate the anti-cancer activity of a crude flavonoid extract derived from N. sativa by using the estrogen receptor–positive breast cancer cell line MCF-7 as a model, as well as to elucidate the molecular mechanism of action. To our knowledge, no study in the literature thus far has elucidated the anti–breast cancer properties of N. sativa flavonoids.
Herbal material and extract preparation
Fresh black seeds (N. sativa) were purchased from the local market of Jeddah, KSA. They were properly washed, thinly grated and used to prepare the extract. 500 gram powder were subjected to maceration in 75% ethanol for seven days, filtered and dried under vacuum. Then, the concentrated extract was defatted by using pet ether. The aqueous portion was then separated and collected. The aqueous portion was then fractionated with N-butanol saturated water. The N-butanol portion was then separated and collected. The aqueous portion was discarded and the N-butanol portion was fractionated with 1% KOH. The 1% KOH portion was then separated, collected and dried. The N-butanol portion will be discarded. The KOH portion was then fractionated with dilute HCl and N-butanol saturated water and the N-butanol portion was separated and collected. The dilute HCl portion was discarded and the N-butanol portion (crude flavonoids fraction) was dried in water bath. The dried crude flavonoid extract of N. sativa (CFENS) was diluted in DMSO to obtain desired concentrations and applied to MCF-7 cells in cultures.
Cell culture and treatments
The human breast cancer cell line, MCF-7, was obtained from King Fahed Biomedical Research Center, King Abdul Aziz University, KSA. The cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin antibiotics in tissue culture flasks under a humidifying atmosphere containing 5% CO2 and 95% air at 37 °C. The cells were sub-cultured at 3–4 day interval.
The effect of CFENS on the viability of MCF-7 cells was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) (Cayman Chemicals, USA) assay following the manufacturer's instructions. Early log phase cells were trypsinized and re-grown in 96-well cell culture plates at the concentration of 104 cells/mL in 100 µL of complete culture medium. Twenty-four hours later, the medium was removed and replaced with fresh medium with or without CFENS. Then, MTT was added to each well and incubated for 4 h after which the plates were centrifuged at 1,800 rpm for 5 min at 4°C. After careful removal of the medium, 0.1 mL of PBS was added to each well and the plates were shaken. The absorbance of converted dye is measured at a wavelength of 570 nm and the increased absorbance is directly proportional to cell viability. The effects of CFENS on growth inhibition were assessed as percent cell viability, where DMSO water-treated cells were taken as 100% viable. For these studies, all experiments were repeated three or more times.
Colonogenic assay
This assay measures the ability of tumor cells to grow and form foci in a manner unrestricted by growth contact inhibition as is characteristically found in normal, untransformed cells. Approximately 500 cells were seeded into six-well plates in triplicate and allowed to adhere overnight. Thereafter, cell culture medium will be changed and cells will be treated with increasing doses of CFENS. The cells were allowed to incubate at 37°C in the incubator undisturbed for 15 days. During this period each individual surviving cell would proliferate and form colonies. On day 15, the colonies were washed with cold phosphate buffer saline, fixed with cold 70% ethanol and stained with 0.25% Giemsa stain.
Apoptotic assay
The nuclear morphological changes associated with apoptosis were analyzed using Hoechst 33342 or acridine orange/ethidium bromide staining as previously described [31]. MCF-7 cells were suspended at a final concentration of 150×103 cells/mL in 24-well plates and allowed to adhere to the bottom of the wells for 24 h before CFENS treatment. Cells were then exposed to indicated concentrations of the CFENS for 24 h, before being washed with PBS and stained with Hoechst 33342 or acridine orange/ethidium bromide for 15 min at 37°C. Subsequently, cells were washed with PBS and viewed under inverted fluorescence microscope (Leica, Germany).
Annexin V-FITC assay for assessment of apoptosis
CFENS-induced apoptosis in MCF-7 cells was determined by flow cytometry using the Annexin V-FITC Apoptosis Detection Kit (Sigma) following the instructions of the manufacturer and as previously detailed [31]. Briefly, after treatment of MCF-7 (300 x 103) cells with CFENS for 24 h, the cells were harvested, resuspended in 500 µL 1X binding buffer, stained with 5 μl of Annexin V-FITC and 10 μl of propidium iodide and incubated for 15 min in the dark at room temperature. Then, the cells were analyzed by flow cytometry using NovoCyte™ Flow Cytometer (Acea).
Determination of ROS production
The generation of ROS was assessed by utilizing DCFH-DA (Cayman Chemicals, USA). Briefly, the MCF-7 cells were cultured in 96-well plates and treated with indicated concentrations of CFENS for 24 h. Then cells were labelled with DCFH-DA for 30 min and washed with PBS. Fluorescence intensity was measured (ex: 485/20 nm and em: 528/20 nm) using a micro-plate reader (BioTek Synergy) and visualized using fluorescent microscope (Leica, Germany).
Determination of intracellular GSH
Intracellular GSH level of HepG2C8 cells was measured with Glutathione Assay Kit (Cayman Chemicals, USA). Briefly, the MCF-7 cells (80 x 103) were cultured in 12-well plates and treated with indicated concentrations of CFENS for 24 h. Then cells were collected and handled following manufacturer’s instructions. The absorbance was measured at 405 nm using a micro-plate reader (BioTek Synergy).
Analysis of lysosomal membrane potential
For lysosomal membrane permeabilization (LMP), the MCF-7 cells were seed and treated as described above. Then, cells were washed and stained with AO for 15 min at 37° C, washed with PBS. Fluorescence intensity was measured (ex: 485/20 nm and em: 528/20 nm) using a micro-plate reader (BioTek Synergy).and visualized using fluorescent microscopy (Leica, Germany).
Analysis of mitochondrial membrane potential
Mitochondrial membrane potential (Δψm) was assessed utilizing a cytofluorimeteric, lipophilic cationic dye, JC-1 (Cayman Chemicals, USA), following manufacturer's recommendations. Briefly, MCF-7 cells were cultured in 96-well plate (10x103 cells/well) and treated with displayed concentrations of CFENS for 24 hours. The cells were labelled with JC-1 dye for 30 min and washed with PBS. Fluorescence intensity was measured (ex: 485/20 nm and em: 528/20 nm) using a micro-plate reader (BioTek Synergy).and visualized using fluorescent microscopy (Leica, Germany).
Detection of activity of caspases
Caspase-3/7, caspase-9 and caspase-8 activities were determined by using the Caspase-Glo® 3/7, Caspase-Glo® 9 and Caspase-Glo® 8 Assay kits, respectively (Promega). The MCF-7 cells were seeded in 100 μL medium in 96-well plates (104 cells/well) and treated with indicated concentrations of CFENS for 24 h. Then, activities of Caspase-3/7 were measured according the manufacturer’s instructions. Briefly, 100 μL of the assay reagents were added to each well and the contents of the wells were mixed using a plate shaker at 300-500 rpm for 3 h. The luminescence of each sample was measured using plate-reading luminometer (BioTek Synergy).
Comet assay (Single Cell Gel Electrophoresis)
CFENS-induced DNA damage was determined using the comet assay. Cells were treated with indicated concentrations of CFENS for 24 h in complete medium, harvested, resuspended in ice-cold PBS and processed under a dimmed light as described earlier [31]. Prepared comet slides were viewed and nuclei images were visualized and captured at 100× magnification with an Axioplan 2 fluorescence microscope (Zeiss) equipped with a CCD camera (Optronics, Goleta, CA, USA).
DNA fragmentation assay
DNA gel electrophoresis was used to determine the presence of internucleosomal DNA cleavage. Briefly, the MCF-7 cells (106 cells/100 mm dish) treated with various concentrations of CFENS for indicated periods, collected, washed in PBS and resuspended in lysis buffer (0.5% Triton X-100 in10 mM EDTA, and 10 mM Tris-HCl, pH 8.0) on ice for 30 min. Then genomioc DNA was extracted as previously detailed [31]. The DNA was then resolved by electrophoresis on 2% agarose gel. After electrophoresis at 100 V, the gel was stained with ethidium bromide, and DNA was visualized by a UV transilluminator (BIO-RAD).
Cell cycle analysis
CFENS-induced cell cycle arrest in MCF-7 cells was determined by flow cytometry analyses. Briefly, cells (300 x 103) were seeded in 25 mL flask overnight; then cells were treated with indicated concentrations of CFENS for 24 h, harvested, washed and fixed in 2 mL of ice-cold 70% ethanol overnight at 4 °C. After fixation, cells were washed, resuspended in 500 µL PBS, incubated with RNase A (20 μg/mL final concentration) and propidium iodide (50 μg/mL final concentration) for 30 min in the dark at room temperature and finally subjected to flow cytometry analysis using NovoCyte™ Flow Cytometer (Acea).
Western blot analysis
For Western blot analysis, polyacrylamide gel electrophoresis and immunoblotting were performed as previously described [31].
Statistical analyses
All experiments were repeated from three to five times independently and in triplicate. In all experiments, the results were presented as mean ± standard deviation (SD) for continuous variables. The statistical significance of results was determined by Student’s t-test, and a p value of less than 0.05 was considered statistically significant.
CFENS inhibits proliferation and clonogenicity in MCF-7 cells
As a basis for investigating the effect of CFENS on cell viability, MCF-7 cells were exposed to increasing concentrations of CFENS (50, 100, 150, and 200 µg/mL), and the inhibition of cell proliferation was determined by an MTT assay. The CFENS treatments achieved a time- and dose-dependent decrease in cell proliferation in as early as 24 h, and this continued up to 72 h of exposure (Figure 1A). The cell viabilities at 48 h and 72 h were found to have minute differences, implying that the cells respond to CFENS within 48 h. The CFENS treatments induced a decrease in MCF-7 cell viability ranging from 88 to 45% after 24 h, from 65 to 25% after 48 h, and from 53 to 17% after 72 h. Half maximal inhibitory concentration (IC50) values are commonly used to evaluate the potency of a compound: the lower the IC50 value, the more potent the compound. According to the results obtained from the MTT assay, the IC50 values were 170, 110, and 45 mg/ml at 24, 48, and 72 h, respectively. To confirm these results, the above experiments were repeated by using the human liver and cervical cancer cell lines HepG2 and HeLa, respectively. CFENS was observed to recapitulate its growth inhibitory potential in the context of both cell lines (Figure 1A). Interestingly, the HeLa cells were the most sensitive to CFENS, and the HepG2 cells were the least sensitive.
Next, the antiproliferative potential of CFENS on MCF-7 cells was determined and verified by using an anchorage-dependent colony formation assay (clonogenicity). As shown in Figure 1B, treatment with 50, 100, 150, and 200 μg/mL of CFENS decreased the colony formation and size in MCF-7 cells. Taken together, these findings indicate that CFENS has an apparent potential to inhibit the growth of MCF-7 cells.
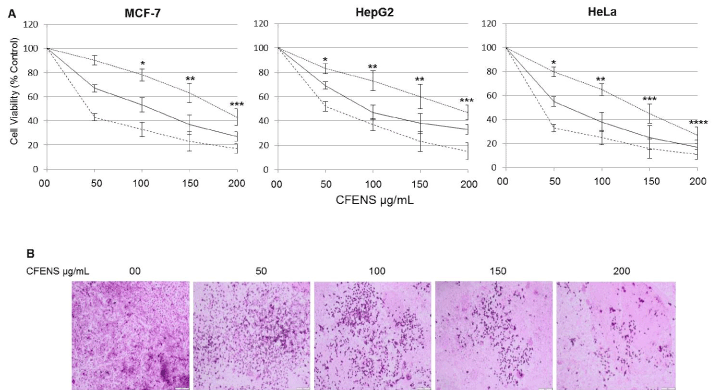
Figure 1. CFENS inhibits proliferation and clongenicity of MCF-7 cells. (A) The displayed cell lines were seeded, at a density of 104/well in 96 wells plates and treated with the indicated concentrations of CFENS for 24 (dotted line), 48 (solid line) and 72h (dashed line). The inhibition of cell proliferation was assessed by the MTT as detailed in section Materials and methods. The experiments were repeated five times in triplicates, and cell viabilities at each dose of the extract were expressed in terms of percent of control and reported as the mean ± SD; (B) MCF-7 cells were seeded onto a 6-well plate at 1000 cells/well and treated with the indicated concentrations of CFENS and assayed as has been detailed in Materials and methods.
CFENS induces apoptotic cell death in MCF-7 cells
Because cell death may proceed through either the apoptotic or the necrotic pathway, the mode of cell death by CFENS was investigated. To test whether the decrease in cell viability observed after treatment with CFENS was due to apoptosis, the treated cells were investigated directly under a microscope. As shown in Figure 2A, the apoptotic morphologic features of cells treated with CFENS appeared in a dose-dependent manner, where the cells showed cellular shrinkage, and at a higher dose of CFENS (150 μg/mL), the majority of the cells detached from the substratum (Figure 2A). Other characteristics of apoptosis, such as membrane blebbing, chromatin condensation, nuclear fragmentation, and formation of apoptotic bodies, were also observed.
Double staining with a mixture of ethidium bromide and acridine orange was used to visualize the viable and the apoptotic cells. As shown in Figure 2, the cells in the control groups showed large green nuclei when observed under a microscope. On the other hand, there was a clear dose-dependent correlation between CFENS treatment and cell apoptosis, characterized by the gradual appearance of orange and red nuclei, which are archetypal signs of apoptotic cells.
Next, the effect of CFENS on the nuclear morphology during cell apoptosis was investigated by using the nuclear stain Hoechst 33342. The nuclei of untreated control MCF-7 cells were stained a less-bright blue and a homogeneous color. In contrast, after treatment with CFENS for 48 h, most cells showed very intense staining of bright and condensed chromatin (Figure 2).
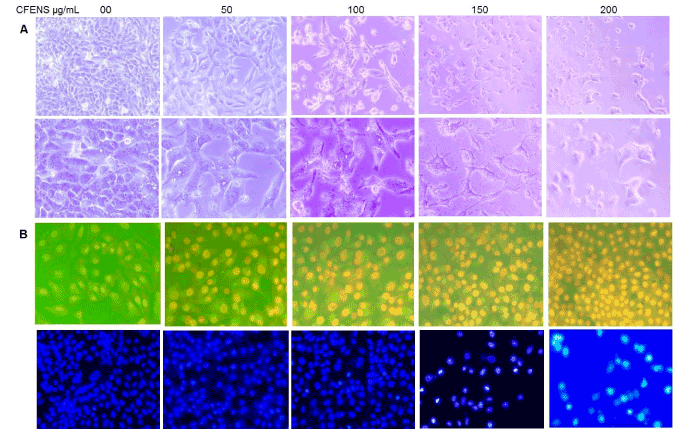
Figure 2. CFENS induces morphological features of apoptosis in MCF-7 cells. The cells were treated with the indicated concentrations of CFENS for 24 h, after which cells were examined for emergence of apoptotic hallmarks. (A) Light microscopy photomicrographs showing morphological changes in MCF-7 cells after incubation with CFENS. Notice cellular shrinkage, detachment and irregularity of cell shape; magnification: 10x (I), 20x (II), 40x (III); (B) Cells stained with 1 μg/mL acridine orange/ethidium bromide for 15 min at 37° C and visualized by florescent microscope. Notice, control cells appear uniformly green, early apoptotic cells with intact plasma membranes appear green, middle stage of apoptosis cells are stained bright green-orange because plasma membrane losses its integrity allowing ethidium bromide enter the cell and at late stage of apoptosis late apoptotic/necrotic nonviable cells are stained red; 20x; (C) Cells were stained with vital nuclear stain (Hoechst 33342) for 15 min at 37°C and visualized by florescent microscope. Note control cells presented homogeneously stained green nuclei of chromatin while nuclei of treated cells stained bright green, indicating chromatin condensation; magnification 20x.
To obtain further proof that CFENS has pro-apoptotic activities, MCF-7 cells were incubated with different concentrations of CFENS for 24 h and then analyzed by flow cytometry by using Annexin V FITC and propidium iodide (PI).
As shown in Figure 3, the CFENS treatments resulted in a significant dose-dependent increase in the number of apoptotic cells at both the early and late stages of apoptosis: 0 µg/mL (control, 9.41%), 100 µg/mL (34.71%), and 150 µg/mL (41.6%). These results indicate that the CFENS treatments significantly induced apoptosis in MCF-7 cells.
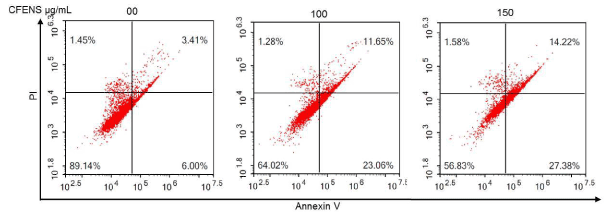
Figure 3. CFENS promotes early and late apoptosis in MCF-7 cells. MCF-7 cells were treated with the displayed concentrations of CFENS, harvested, stained with annexin V/PI, and analyzed by flow cytometry.
CFENS induces ROS accumulation, GSH depletion, and permeabilization of lysosomal and mitochondrial membranes in MCF-7 cells
Most phytochemical agents induce apoptosis through the induction of ROS [19]. To determine whether the pro-apoptotic potential of CFENS is mediated through ROS accumulation, MCF-7 cells were incubated with DCFH-DA stain, an indicator of peroxide and superoxide accumulation. As shown in Figure 4A, the quantitative analyses showed a dose-dependent increase in ROS generation after CFENS treatment. Next, the effect of CFENS on ROS production was monitored by using a fluorescence microscope. The fluorescent images showed that the intensity of green fluorescence of DCFH-DA increased consistently with increasing doses of CFENS. These results indicate that CFENS caused an increase in ROS level.
When cells suffer increased levels of oxidative stress, the intracellular GSH level is depleted, whereas GSSG is accumulated, resulting in a decrease in the GSH/GSSG ratio. To find out whether CFENS is able to deplete the intracellular GSH level, the level of GSH after CFENS treatment was determined. The findings shown in Figure 4B indicate that CFENS treatment remarkably and dose-dependently depleted the intracellular GSH level. The GSH levels were significantly decreased by 60%, 75%, and 80%, respectively, with 100, 150, and 200 μg/mL of CFENS. Collectively, these data support the hypothesis that CFENS can induce oxidative stress in MCF-7 cells.
One of the earliest intracellular events that occur after ROS accumulation is lysosomal membrane permeabilization (LMP), which leads to the discharge of lysosomal enzymes into the cytosol. These enzymes are capable of signaling the mitochondrial-mediated pathway of apoptotic death. To determine whether CFENS-induced ROS accumulation could trigger a disruption of the lysosomal membrane, cells were treated with CFENS and stained with AO. AO is a lysosomotropic fluorochrome that preferentially accumulates in lysosomes, at high concentration, and emits red fluorescence; however, when AO is present in the cytosol and nucleus, for example, after LMP, green fluorescence is emitted. The spectrophotometric analysis of AO fluorescence presented in Figure 4C shows a gradual increase in green fluorescence, indicating rupture of the lysosomal membrane and relocation of acridine orange from the lysosomes to the cytoplasm. Consistent with the results of the spectrophotometry, the fluorescent images in Figure 4C show that CFENS caused a marked decrease in red fluorescence and an increase in cytosolic green fluorescence. These observations provide supporting evidence that the lysosomal membrane underwent permeabilization, leading to the release of lysosomal hydrolytic enzymes into the cytosol.
ROS accumulation also induces outer mitochondrial membrane permeabilization (MMP), which leads to loss of mitochondrial membrane potential (ΔΨm) and the release of pro-apoptotic factors into the cytosol. To assess whether CFENS triggers MMP, the effect of CFENS on mitochondrial membrane potential was monitored by using JC-1 dye. In viable cells, the JC-1 dye accumulates in mitochondria in aggregate forms and emits red fluorescence. However, when mitochondrial potential is lost due to apoptotic cell death, JC-1 aggregates are released into the cytosol as monomers and emit green fluorescence. Thus, cells were treated with CFENS and labeled with JC-1 dye. Then, the ratio of green to red fluorescence was quantified by using a microplate reader. As shown in Figure 4E, the ratio of red to green fluorescence markedly decreased in a dose-dependent manner. The effects of CFENS on MMP were also examined by using a fluorescence microscope after staining with JC-1. As shown in Figure 4D, the intensity of red fluorescence decreased with increasing doses of CFENS, whereas the intensity of green fluorescence increased. These observations indicate that CFENS efficiently induced MMP, leading to the release of JC-1 aggregates and their distribution as monomers in cytosol.
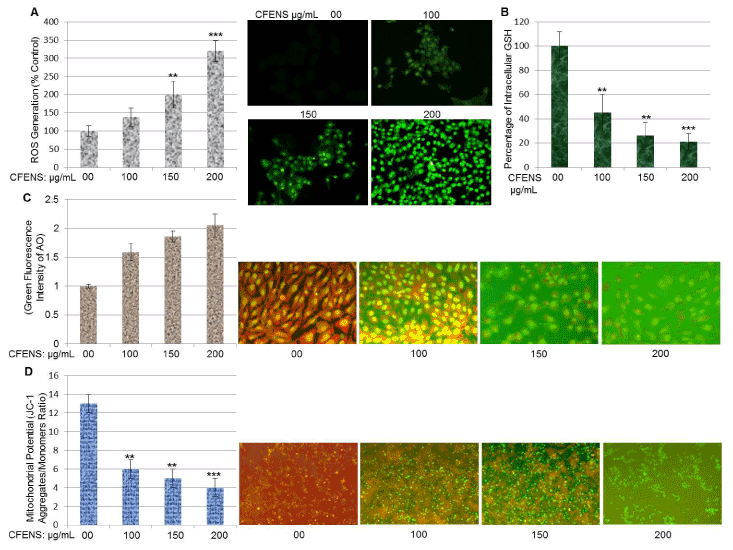
Figure 4. CFENS induces ROS accumulation, GSH depletion and mitochondrial membrane depolarization in MCF-7 cells. (A) Cells were treated with indicated concentrations of CFENS for 24 h; then stained with DCFH-DA and the DCF fluorescence intensity was measured by a fluorescence spectrophotometer and fluorescent microscopy. (B) Cells were treated with indicated concentrations of CFENS for 24 h and assayed for GSH levels as detailed in Materials and Methods. (C) Cells were treated with indicated concentrations of CFENS for 24 h; then stained with acridine orange (AO) the AO fluorescence intensity was measured by a fluorescence spectrophotometer and fluorescent microscopy. (D) Cells were treated with the indicated concentrations of CFENS for 24. Then, cells were stained with a mitochondria-specific dye, JC-1, and its florescence was monitored using fluorescence microscopy. Note, JC-1 stain is accumulated in control cells, where it displayed a bright-red fluorescence indicating a high potential. In contrast, JC-1 stain is poorly accumulated in CFENS-treated cells, which display green fluorescence, indicating the loss of the mitochondrial membrane potential.
CFENS increases the Bax/Bcl-2 ratio and induces activation of caspases-3/7 and 9 in MCF-7 cells
Molecularly, the MMP is controlled by Bcl-2 family proteins. The key members of this family are the pro-apoptotic protein Bax and the anti-apoptotic protein Bcl-2. The ratio of Bax/Bcl-2 expression determines whether a cell resumes survival or commits apoptotic death. To investigate whether CFENS-induced MMP is related to alteration of the Bax/Bcl-2 ratio, cells were treated with CFENS, and the expression levels of both proteins were monitored by using Western blot analyses. The results shown in Figure 5A indicate that CFENS consistently affected the expression levels of both proteins. Although it upregulated the expression of the Bax protein, it downregulated the expression of the Bcl-2 protein, leading to an increase in the Bax/Bcl-2 ratio. All these observations are specific to the CFENS effect because, when the Western blot was re-probed for β-actin, there was equal loading of proteins in all treatments.
An increase in the Bax/Bcl-2 ratio leads to the release of proapoptotic factors into the cytosol, with subsequent activation of the apoptotic machinery. The central components of this machinery are the caspases of a proteolytic system, including initiator caspases (caspases-8 and -9) and effector caspases (caspase-3/7), which play a central role during the executional phase of apoptosis. As shown in Figure 5B, the exposure of MCF-7 cells to CFENS for 24 h resulted in a dose-dependent increase in caspase-3/7. These are significant results because caspase-3/7 activation represents the irreversible or execution stage of apoptosis. When the cells were incubated with 100, 150, and 200 μg/mL, the caspase-3/7 activity was markedly increased by 6.5, 8.3, and 9%, respectively, compared with the control cells. In addition, the CFENS treatments activated caspase-9 and, to a much lesser extent, caspase-8, indicating that the major signaling pathway induced by CFENS is the mitochondrial one.
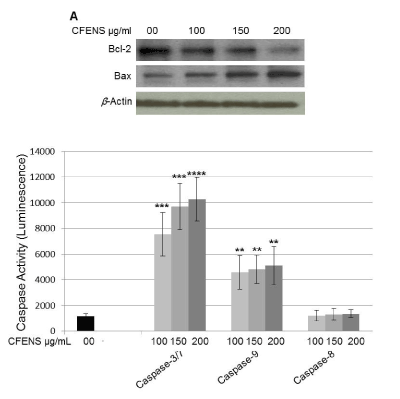
Figure 5. CFENS modulates expression levels of Bcl-2 and Bax proteins and induces activation of caspases 3/7 and 9 in MCF-7 cells. (A) (CFENS treatment up-regulated expression of the Bax protein and down-regulated expression of the Bcl-2 protein. The MCF-7 cells were incubated with the indicated concentrations of the CFENS for 24 h; then total protein lysates were prepared and subjected for Western analyses. The experiments were repeated several times and typical results from independent experiments are shown. (B) MCF-7 cells were seeded on a 96-well luminometer plate (104 cells/well) and treated with indicated concentrations of CFENS for 24 h. Then, activities of Caspases 3/7, 9 and 8 were evaluated as detailed in Materials and Methods.
CFENS induces DNA damage lacking oligo-nucleosomal degradation
A growing body of evidence shows that ROS generation induces DNA damage, including a multitude of oxidized base lesions, abasic sites, and single- and double-strand breaks, all of which, if incapable of repair, can lead to the onset of cytotoxic and/or mutagenic apoptotic events [32]. To confirm whether the CFENS treatments induced DNA damage, a comet assay was carried out. This assay is a sensitive method for monitoring single-strand DNA breaks at the single-cell level and is used as a biomarker of apoptosis. As shown in Figure 6A, untreated control MCF-7 cells had no detectable comet tails. On the other hand, incubation of cells with 100 μg/mL CFENS resulted in the appearance of comet tails in a dose-dependent manner. These results indicate that CFENS treatment markedly induced DNA damage in MCF-7 cells.
The degradation of DNA into oligo-nucleosomal fragments of multiples of 180 base pairs is one of the essential features of classic apoptotic cell death. Therefore, we examined whether CFENS might induce DNA fragmentation in MCF-7 cells. Figure 6B shows that there was no DNA fragmentation after treatment with CFENS for 72 h. Therefore, we concluded that CFENS uses a non-classic DNA fragmentation scenario to process chromatin during apoptosis.
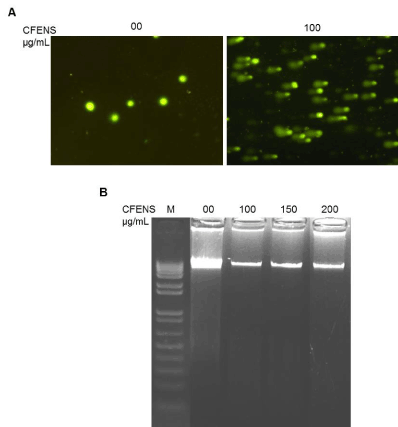
Figure 6. CFENS induces DNA damage lacking oligo-nucleosomal degradation. MCF-7 cells were treated with the indicated concentration of CFENS for 48 h; then harvested and assayed (A) Comet assay showing DNA damage in MCF-7 6 cells treated with CFENS; magnification 10X. Notice treated cells show clear appearance of a fan like comet formation, which is a typical characteristic of apoptotic phenomenon; (B) Agarose gel electrophoresis shows CFENS treatment did not induce oligo-nucleosomal degradation of the genomic DNA. Lane `M’ indicates the DNA marker ladder.
CFENS induces cell cycle arrest and modulates the expression of cell cycle-regulating proteins
Because cell proliferation is closely associated with cell cycle progression, and CFENS inhibited the proliferation of MCF-7 cells, the possible inhibitory effect of CFENS on the progression of the MCF-7 cell cycle was examined. As shown in Figure 7A, exposure of MCF-7 cells to 100 and 150 μg/mL CFENS for 24 h resulted in the accumulation of cells in the G1/G0 phase, whereas treatment of cells with 150 μg/mL CFENS caused ~1.7-fold enrichment of cells in the G1/G0 phase, accompanied by a 15% and 30% decrease, respectively, in the S and G2/M phases. These data suggest that the inhibition of cell proliferation or the induction of cell death in MCF-7 cells by CFENS may be associated with the induction of G1/G0 phase arrest. To find an indication of the mechanisms involved in cell cycle arrest, we investigated whether CFENS could affect the expression levels of p53, p21, and cyclin D1 proteins, which play central roles in cell cycle progression [33]. The cells were incubated with increasing concentrations of CFENS for 24 h. Then, total protein lysates were prepared and subjected to Western blot analyses. As shown in Fig. 7B, treatment with CFENS markedly increased the level of p53 and p21 proteins, accompanied by a decrease in the expression of cyclin D1. Collectively, these data suggest that CFENS induced cell cycle arrest and modulated the expression levels of the cell cycle regulatory proteins p53, p21, and cyclin D1 in a manner that contributed to the susceptibility of MCF-7 cells to CFENS-induced apoptosis/inhibition of growth.
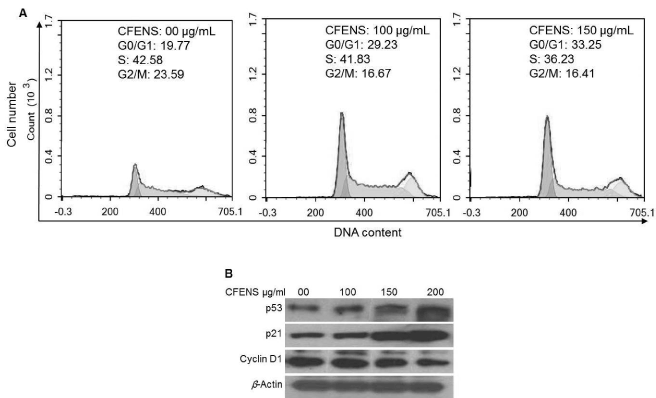
Figure 7. CFENS induces cell cycle arrest and modulates expression of cell cycle-regulating proteins in MCF-7 cells. (A) CFENS induces G1/G0 phase arrest Cells were incubated with the displayed concentrations of CFENS for 24 h, harvested, labelled with propidium iodide and analyzed by flow cytometry. (B) The MCF-7 cells were incubated with the indicated concentrations of the CFENS for 24 h; then total protein lysates were prepared and subjected for Western analyses. CFENS treatments modulate up-regulated expression of p53 and p21 proteins and down-regulated expression of cyclin D1 protein. The experiments were repeated several times and typical results from independent experiments are shown.
Breast cancer endangers the lives of women worldwide. Approximately 75% of breast cancers are classified as estrogen receptor–positive. This phenotype is generally driven by the estrogen/estrogen receptor pathway, and endocrine therapies targeting this pathway are the pillars for managing this disease. However, these endocrine-based therapies are hampered by the development of biological resistance, which leads to disease progression and eventual death. In fact, the problem of drug resistance is clearly evident in most of the current chemotherapeutics that apply a single-molecule, single-target, and single-drug strategy. Recently, the Food and Drug Administration (FDA) endorsed a combination therapy strategy for cancer [34,35]. This combination therapy could be accomplished through the use of medicinal plants; there is convincing evidence that medicinal plants could supply novel bioactive molecules that could target, in an additive or synergistic manner, multiple signaling pathways in cancer cells [36]. Therefore, the present research investigated the effect of crude flavonoid extracts derived from N. sativa seeds on the growth of breast cancer MCF-7 cells. We hypothesized that the bioactive constituents in a crude flavonoid extract formula might additively or synergistically target multiple pathways or multiple targets in a single pathway boosting MCF-7 cell growth. Coherent to our rationale, CFENS consistently suppressed the growth and clonogenicity of the MCF-7 cells in a dose- and time-dependent manner. Furthermore, CFENS inhibited the growth of the hepatocellular and cervical carcinoma cell lines HepG2 and HeLa cells, respectively. Therefore, this study identified a novel dietary agent, CFENS, as a putative agent to control the growth of breast cancer.
Most chemotherapeutic agents derived and developed from natural products exert their anticarcinogenic activities by the induction of apoptosis in breast and other cancer cells [15,37]. Similarly, the present work found that CFENS inhibited the growth of MCF-7 cells by signaling apoptotic cascades. This is because typical morphologic hallmarks of apoptotic death [22], such as loss of cell viability, cellular shrinkage, irregular cell shapes, and sporadic distribution, were observed after MCF-7 cells were exposed to CFENS. On the other hand, untreated cells appeared as healthy intact monolayers with a regular distribution of typical fibroblast-like–shaped cells. The findings with the use of the fluorescent stains AO/EtBr are parallel to the observations under a light microscope. Basically, the importance of these stains is that only AO is membrane-permeable dye that can be ingested by viable cells and fluoresces green fluorescence. On the other hand, EtBr is a membrane-impermeable dye; it can be ingested only by late apoptotic/necrotic cells, and it stains the nuclei of cells red. When both dyes enter cells (e.g., at late apoptotic stage/necrotic death), the cells appear red because the red fluorescence of EtBr dominates the green fluorescence of AO. At early apoptotic stage, the cells maintain their membrane integrity; accordingly, when viable cells and/or early apoptotic cells are stained with AO/EtBr, they appear green. On the other hand, when cells undergo apoptosis, they lose their membrane integrity, leading to EtBr entry and binding to DNA, which stains cells orange/red. Therefore, dual staining of cells with a mixture of AO and EtBr can distinguish viable, early apoptotic, and late apoptotic cells. Interestingly, when untreated MCF-7 cells were stained with AO/EtBr, all cells appeared green, indicating that the cells were viable and had intact membranes. On the other hand, when cells were treated with 75 µg/ml CFENS, some cells showed red nuclei, indicating that they had lost their membrane integrity, thus allowing EtBr entry and binding to DNA. Further increasing the CFENS doses increased the number of cells with red nuclei, indicating a specific cytotoxic effect of CFENS. At the highest concentration of CFENS (200 µg/ml), all cells showed intense red nuclei, indicating that all cells underwent events of late apoptosis/necrosis. In addition, when untreated cells were labeled with Hoechst 33342 dye, another widely used stain for detection of apoptosis, they showed a homogenous blue color. Meanwhile, treated cells showed a bright blue color, indicating chromatin condensation; with a further increase in the CFENS doses, some cells showed nuclear fragmentation, which is a hallmark of apoptotic nuclei. Thus, there is an overlap among all the morphologic assays (light microscopy and staining with AO/EtBr and Hoechst 33342), indicating that the growth inhibitory potential of CFENS is related to its competency to initiate apoptotic cascades in MCF-7 cells.
Next, we illuminated the molecular mechanism mediating the apoptogenic potentiality of CFENS. Accumulative evidence indicates that chemotherapeutics and phytochemicals induce apoptosis, in part through the generation of ROS and the disruption of redox homeostasis [38-40]. Within the physiologic circumstances, the generation of excessive ROS is downregulated by nonprotein antioxidants, such as GSH, as well as by an antioxidant enzyme, such as catalase [25]. Our results indicate that there was a consistent and monotonic negative linear correlation between the ROS and GSH levels after the MCF-7 cells were treated with CFENS; a dramatic ROS burst and a noticeable decrease in the GSH level was observed. Because the generation of ROS (oxidant) and the depletion of GSH (antioxidant) per se are typical intracellular conditions that promote the initiation of apoptotic cascades, it is tempting to speculate that the CFENS-initiated oxidative stress status provoked apoptotic cascades in MCF-7 cells.
Among the known consequences of ROS accumulation is the induction of LMP, an event that ultimately leads to the release of lysosomal hydrolytic enzymes into cytoplasm. Such release is a potentially lethal event because these enzymes can unselectively degrade nearly all cellular components and can also directly activate procaspase-3 [41]. Indeed, mounting evidence indicates that many putative cancer chemopreventive agents can signal apoptotic death through the lysosomal apoptotic pathway [42,43]. Therefore, we investigated whether CFENS may lysosomal apoptotic pathway in MCF-7 cells. To this end, the lysosomal membrane integrity was determined by using AO. This dye preferentially accumulates in lysosomes and fluoresces red, but it emits a green fluorescence when presented in the cytoplasm/nucleus. Based on the fluorescent images, untreated MCF-7 cells showed a red fluorescence confined to the lysosomals. On the other hand, after treatment with CFENS, lysosomal AO was discharged into the cytosol. Furthermore, the spectrophotometry analysis showed that treatment of MCF-7 cells with CFENS led to a dose-dependent increase of green fluorescence. These findings indicate that the CFENS treatments induced LMP in MCF-7 cells, which may promote downstream cascades (e.g., activation of caspase-3), leading to the initiation of apoptosis. Another corollary related to CFENS-induced LMP is the induction of MMP, in which the released lysosomal enzymes could promote MMP loss and the subsequent release of mitochondrial proapoptotic factors [44].
Exponential studies 2021 Copyright OAT. All rights reservediate the initiation of mitochondria-dependent apoptotic cascades through the induction of ROS accumulation [19]. In fact, ROS accumulation induces the collapse of mitochondrial membrane potential, causing MMP, which is one of the earliest intracellular events of apoptosis [24,45]. However, mitochondrial potential is controlled by members of Bcl-2 family proteins. This protein family embraces two main subfamilies, which include antiapoptotic members (such as Bcl-2) and antiapoptotic members, such as Bax [46,47]. The Bcl-2 protein inhibits apoptosis by preserving the mitochondrial membrane integrity, which inhibits the release of proapoptotic factors. On the other hand, Bax opposes the Bcl-2 protein function; it induces MMP, leading to the discharge of proapoptotic factors (e.g., cytochrome c and AIF) into the cyctosol, in which they form, along with other cytoplasmic proteins, apoptosomes that initiate the activation of downstream effectors, such as caspase-9 [20]. Thereby, fine-tuning of the Bcl-2 and Bax expression levels is the main factor that controls cell fate: survival versus suicide [47]. To explore the possible apoptotic mechanism of CFENS through the mitochondrial pathway, the impact of CFENS on the mitochondrial integrity and the expression levels of Bcl-2 and Bax were investigated. CFENS was found to cause MMP, as confirmed by a decrease of red fluorescence and an increase of green fluorescence of the JC-1 dye. Furthermore, Western blot analysis showed that CFENS dose-dependently increased the expression level of the Bax protein and lowered that of the Bcl-2 protein, ultimately leading to a noticeable increase in the Bax/Bcl-2 protein ratio compared with the control group. These observations indicate that disruption of the mitochondrial membrane potential and an imbalance in the Bax/Bcl-2 ratio due to CFENS are key events that might participate in triggering apoptotic cascades in MCF-7 cells.
Cumulative studies have explained that most anticancer agents per se could mediate their apoptotic effect directly through the induction of DNA damage [19]. DNA damage is an irreversible event and occurs at late-stage apoptotic episodes [48,49]. To determine whether DNA damage is related to the proapoptotic activity of CFENS, a comet assay was carried out. This assay is widely accepted as a classic marker in studying the apoptotic activity of anticancer agents because it can reliably assess DNA damage at a single-cell level and can distinguish the mode of cell death, whether apoptotic or necrotic [50,51]. Interestingly, our findings indicate that CFENS induced DNA damage, as evidenced by the appearance of comet tails. If a cell with damaged DNA fails to undergo repair, it proceeds to apoptotic death [52]. Thus, CFENS-induced damage of genomic DNA in MCF-7 cells might motivate cells toward apoptotic death.
Earlier studies indicated that apoptotic machinery follows two parallel pathways to process chromatin during apoptosis. One pathway involves the activation of caspases and the eventual oligo-nucleosomal DNA fragmentation. The other pathway mediates the relocation of a mitochondrial protein (AIF) to the nucleus, resulting in peripheral chromatin condensation and large-scale DNA fragmentation, with a size of ~50 kbp [53]. The treatment of MCF-7 cells with CFENS resulted in chromatin condensation, as shown by the vital nuclear stain Hoechst 33342 (Figure 2); however, it did not produce oligo-nucleosomal DNA fragmentation (Figure 6B). Interestingly, in our previous study, the treatment of MCF-7 cells with a crude extract derived from the medicinal herb Rhazya stricta was found to induce oligo-nucleosomal DNA fragmentation [54]. Thus, the failure to detect oligo-nucleosomal DNA fragmentation in response to CFENS is not due to impairment of the apoptotic machinery in MCF-7 cells. Rather, the apoptotic machinery of MCF-7 cells seems to differentially processes the chromatins of apoptotic cells, depending on the constituents and ingredients of the extract used. Therefore, the CFENS-dependent DNA processing machinery applied a noncanonical scenario in processing the chromatins of apoptotic cells. Similarly to CFENS, other chemotherapeutic agents, such as curcumin [55] and matrine [56], have been found to induce apoptosis without oligo-nucleosomal DNA cleavage.
The central player in the apoptotic machinery is a family of proteases named caspases. Basically, these proteins are divided into initiator and executioner caspases [21]. Initiator caspases are the apical mediators in the axis of apoptotic cascades; classically, they are represented by caspases-8 and -9. Caspase-8 is the downstream effector of cell death receptors and is the apical caspase in the external apoptotic pathway, whereas caspase-9 is the downstream mediator of the mitochondrial (internal) apoptotic pathway [21]. Nevertheless, both caspase-8 and -9–dependent pathways converge downstream at caspase-3 [21]. Caspase-3 is a typical executioner caspase of programmed cell death; its activation provokes downstream events leading to cleavage of DNA, protein machineries, and other macromolecules essential for cell survival, which results in the emergence of hallmarks of apoptosis [22]. Many natural compounds have been found to initiate apoptosis in MCF-7 cells through caspase-3 [57]. Similarly, the present results showed that CFENS treatment caused the activation of caspases-3 and -9, adding further proof that CFENS has the potential to trigger events leading to the initiation of the mitochondrial apoptotic pathway.
Molecular analyses of human cancers have indicated that cell cycle regulators are frequently mutated in most common malignancies [33]; hence, control of the cell cycle progression in cancer cells is gaining momentum as an effective strategy to control cancerous growths [58,59]. The observations in this study show that the antiproliferative activity induced by CFENS in MCF-7 cells is linked to its ability to trigger cell cycle arrest. To further confirm this effect, we analyzed the expression levels of p53, p21, and cyclin D1 proteins, which are central players in cell cycle progression. For example, p53 is an onco-suppressor protein and the guardian of the genome; in response to cellular stress leading to DNA damage, the p53 pathway is activated to maintain the integrity of the genome. If p53 signaling does not respond to DNA damage, the cell with DNA damage can turn into cancer cells [60]. When a cell develops oncogenic lesions, p53 is activated and induces cell cycle arrest and, consequently, apoptosis, which prevents the propagation of oncogenic lesions to the descendent progeny [61]; therefore, suppression of p53 activity plays a central role in neoplastic growths. Not surprisingly, many chemopreventive agents have been found to impose their proapoptotic potential by activating p53 signaling pathways [62]. p53 exerts its apoptotic function through both transcription-independent and -dependent mechanisms. In its transcription-independent apoptotic activity, p53 physically interacts with the antiapoptotic Bcl-2 family proteins Bcl-XL and Bcl-2, inhibiting their antiapoptotic function [63]. In its transcription-dependent apoptotic activity, p53 activates the expression of a myriad of genes transcribing proapoptotic proteins (such as Bax, Puma, Noxa, and Fas) or cell cycle arrest proteins (such as p21), as well as other factors [63].
Another finding that deserves attention is the induction of p21 by CFENS, given that p21 is the global inhibitor of cell cycle progression and one of the most important and potent effectors induced by p53 in response to DNA damage [33]. Many dietary agents prevent the growth of various cancerous cell lines through p21-dependent activities [64]. Therefore, it is tempting to suggest that the upregulation of p53 and p21 after CFENS treatments might trigger molecular events leading to apoptosis induction in MCF-7 cells. In addition to its ability to induce cell cycle arrest by increasing the expression level of the universal cell cycle inhibitor p21, p53 can also induce cell cycle arrest by directly inhibiting the activity of cyclins and cyclin-dependent kinases (CDKs) [63]. Cyclins and CDKs are a network of protein complexes that drive cell cycle progression; hence, the deregulated expression of these complexes promotes the development, progression, and drug resistance of breast cancers [65]. For example, cyclin D1 has been shown to cooperate with other oncogenes in the transformation of normal breast cells into malignant phenotypes. Furthermore, the cyclin D1 gene has been found to be amplified in nearly 15 to 20% of patients with estrogen receptor–positive breast cancer, whereas the cyclin D1 protein is overexpressed in approximately 50 to 70% [66]. Besides its ability to drive cell cycle progression through activation of its catalytic partner CDK-4/6, cyclin D1 can also boost the proliferation of breast cancer cells through direct activation of estrogen receptors [67]. Finally, cyclin D1 is also involved in the development of other cancer phenotypes, such as hepatocellular carcinoma; colorectal, lung, and bladder cancers; and some forms of lymphomas and carcinomas [66]. Similarly to CFENS, several phytochemicals, including curcumin, resveratrol, genistein, and apigenin, have been found to prevent breast cancer cell growth through the downregulation of cyclin D1 [68]. These results indicate that CFENS-dependent modulation of the expression of cell cycle regulatory proteins might trigger a series of events leading to cell cycle arrest, which may contribute, at least in part, to suppression of MCF-7 cell growth.
The results of the present study indicate that CFENS inhibits proliferation and induces apoptosis in MCF-7 cells in a dose-dependent manner. The apoptogenic potential of CFENS involves the accumulation of ROS, depletion of GSH depolarization of the mitochondrial membrane, damage of DNA, increase of the Bax/Bcl-2 ratio, and induction of cell cycle arrest. Further downstream of the apoptosis cascade, CFENS activated caspases-9 and -3. Other molecular mechanisms of CFENS entail the downregulation of cyclin D1 and the upregulation of p53 and p21. Further studies on the in vivo activity of CFENS toward MCF-7 xenograft tumors in nude mice are in progress.
This project was funded by the Deanship of Scientific Research (DSR), at King Abdulaziz University, Jeddah, under grant no. 25/130/1433. The authors, therefore, acknowledge with thanks DSR for technical and financial support.
The authors declare that there is no conflict of interest of a scientific or commercial nature. The authors have no relevant affiliations to, or financial support from any organization that may have a financial interest in the subject matter.
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136: E359. [Crossref]
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017; 67: 7-30.
- Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS et al. Molecular portraits of human breast tumours. Nature. 2000; 406: 747-752. [Crossref]
- Hayes EL, Lewis-Wambi JS. Mechanisms of endocrine resistance in breast cancer: an overview of the proposed roles of noncoding RNA. Breast Cancer Res. 2015; 17: 40. [Crossref]
- Stoff-Khalili MA, Dall P, Curiel DT. Gene therapy for carcinoma of the breast. Cancer Gene Ther. 2006; 13:633-647.
- Csermely P, Agoston V, Pongor S. The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol Sci. 2005; 26: 178-182. [Crossref]
- Rodriguez-Casado A. The Health Potential of Fruits and Vegetables Phytochemicals: Notable Examples. Crit Rev Food Sci Nutr. 2014; 56:1097-1107. [Crossref]
- Ahmad A, Husain A, Mujeeb M, Khan SA, Najmi AK, Siddique NA, Damanhouri ZA, Anwar F. A review on therapeutic potential of Nigella sativa: A miracle herb. Asian Pac J Trop Biomed. 2013; 3:337-352. [Crossref]
- Ali BH, Blunden G. Pharmacological and toxicological properties of Nigella sativa. Phytother Res. 2003; 17:299-305. [Crossref]
- Rahmani AH, Alzohairy MA, Khan MA, Aly SM. Therapeutic Implications of Black Seed and Its Constituent Thymoquinone in the Prevention of Cancer through Inactivation and Activation of Molecular Pathways. Evid Based Complement Alternat Med. 2014; 2014:724658. DOI: 10.1155/2014/724658. [Crossref]
- Meziti A, Meziti H, Boudiaf K, Mustapha B, Bouriche H. Polyphenolic profile and antioxidant activities of Nigella sativa seed extracts in vitro and in vivo. Int J Biol Biomol Agr Food Biotechnol Eng. 2012; 6:109-117. [Crossref]
- Farag MA, Gad HA, Heiss AG, Wessjohann LA. Metabolomics driven analysis of six Nigella species seeds via UPLC-qTOF-MS and GC–MS coupled to chemometrics. Food Chem. 2014; 151:333-342. [Crossref]
- Chahar MK, Sharma N, Dobhal MP, Joshi YC. Flavonoids: a versatile source of anticancer drugs. Pharmacogn Rev. 2011; 5:1-12. [Crossref]
- Kozłowska A, Szostak-Węgierek D. Flavonoids: food sources and health benefits. Rocz Panstw Zakl Hig. 2014; 65:79-85. [Crossref]
- Mocanu MM, Nagy P, Szöllosi J. Chemoprevention of breast cancer by dietary polyphenols. Molecules. 2015; 20:22578-22620. [Crossref]
- Kale A, Gawande S, Kotwal S. Cancer phytotherapeutics: role for flavonoids at the cellular level. Phytother Res. 2008; 22: 567-577. [Crossref]
- Ramos S. Effects of dietary flavonoids on apoptotic pathways related to cancer chemoprevention. J Nutr Biochem. 2007; 18:427-442. [Crossref]
- See comment in PubMed Commons belowHanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674. [Crossref]
- Khan N, Afaq F, Mukhtar H. Apoptosis by dietary factors: the suicide solution for delaying cancer growth. Carcinogenesis. 2007; 28:233-239. [Crossref]
- Wong RS. Apoptosis in cancer: from pathogenesis to Treatment. J Exp Clin Cancer Res. 2011; 30:87. DOI: 10.1186/1756-9966-30-87. [Crossref]
- Zimmermann KC, Bonzon C, Green DR. The machinery of programmed cell death. Pharmacol Ther. 2001; 92:57-70. [Crossref]
- Saraste A, Pulkki K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res. 2000; 45:528-537. [Crossref]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007; 87:99-163. [Crossref]
- Fruehauf JP, Meyskens FL Jr. Reactive Oxygen Species: A Breath of Life or Death? Clin Cancer Res. 2007; 13:789-794. [Crossref]
- Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007; 12:913-922. [Crossref]
- Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic Biol Med. 2010; 49:1603-1616. [Crossref]
- Wang J, Yi J. Cancer cell killing via ROS. To increase or decrease, that is the question. Cancer Biol Ther. 2008; 7:1875-1884. [Crossref]
- Lushchak VI. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J Amino Acids. 2012; 2012:736837. DOI: 10.1155/2012/736837. [Crossref]
- Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, Marinari UM, Domenicotti C. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. 2013; 2013:972913. DOI: 10.1155/2013/972913. [Crossref]
- Park WH, Kim SH. MAPK inhibitors augment gallic acid-induced A549 lung cancer cell death through the enhancement of glutathione depletion. Oncol Rep. 2013; 30:513-519. [Crossref]
- Elkady AI, Hussein RA, El-Assouli SM. Mechanism of action of Nigella sativa on human colon cancer cells: the suppression of AP-1 and NF-κB transcription factors and the induction of cytoprotective genes. Asian Pac J Cancer Prev. 2015; 16:7943-7957. [Crossref]
- Paz-Elizur T, Sevilya Z, Leitner-Dagan Y, Elinger D, Roisman LC, Livneh Z. DNA repair of oxidative DNA damage in human carcinogenesis: potential application for cancer risk assessment and prevention. Cancer Lett. 2008; 266:60-72. [Crossref]
- Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003; 36:131-149. [Crossref]
- Verpoorte R. Good practices: the basis for evidence-based medicines. J Ethnopharmacol. 2012; 140:455-457.
- Dolgin E. When it takes two to tango FDA suggests a new regulatory dance. Nat Med. 2011; 17:270. DOI: 10.1038/nm0311-270. [Crossref]
- Liu, RH. Potential synergy of phytochemicals in cancer prevention: mechanism of action. J Nutr. 2004; 134:3479S-3485S. [Crossref]
- Safarzadeh E, Shotorbani S, Baradaran B. Herbal medicine as inducers of apoptosis in cancer treatment. Adv Pharm Bull. 2014; 4:421-427. [Crossref]
- Martin KR. Targeting apoptosis with dietary bioactive agents. Exp Biol Med. 2006; 231:117-129. [Crossref]
- Antosiewicz J, Ziolkowski W, Kar S, Powolny, AA, Singh SV. Role of reactive oxygen intermediates in cellular responses to dietary cancer chemopreventive agents. Planta Med. 2008; 74:1570-1579. [Crossref]
- Millimouno FM, Dong J, Yang L, Li J, Li X. Targeting apoptosis pathways in cancer and perspectives with natural compounds from mother nature. Cancer Prev Res, 2014; 7:1081-1107. [Crossref]
- Hishita T, Tada-Oikawa, S, Tohyama, K, Miura, Y, Nishihara, T, Tohyama, Y, Yoshida, Y, Uchiyama, T, Kawanishi S. Caspase-3 activation by lysosomal enzymes in cytochrome c-independent apoptosis in myelodysplastic syndrome-derived cell line P39. Cancer Res. 2001; 61:2878-2884. [Crossref]
- Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008; 27:6434–6451. [Crossref]
- Česen MH, Pegan K, Spes A, Turk B. Lysosomal pathways to cell death and their therapeutic applications. Exp Cell Res. 2012; 318:1245-1251. [Crossref]
- Zhao M, Antunes F, Eaton JW, Brunk UT. Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur J Biochem. 2003; 270:3778–3786. [Crossref]
- Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001; 15:2922-2933. [Crossref]
- Cory S and Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002; 2:647-656. [Crossref]
- Del Poeta G, Venditti A, Del Principe MI, Maurillo L, Buccisano F, Tamburini A, Cox MC, Franchi A, Bruno A, Mazzone C, Panetta P, Suppo G, Masi M, Amadori S. Amount of spontaneous apoptosis detected by Bax/Bcl-2 ratio predicts outcome in acute myeloid leukemia (AML). Blood. 2003; 101:2125-2131. [Crossref]
- Norbury CJ and Zhivotovsky B. DNA damage-induced apoptosis. Oncogene. 2004; 23:2797-2808. [Crossref]
- Collins JA, Schandi CA, Young KK, Vesely J, Willingham MC. Major DNA fragmentation is a late event in apoptosis. J Histochem Cytochem. 1997; 45:923-934. [Crossref]
- Archana M, Bastian, Yogesh TL, Kumaraswamy KL. Various methods available for detection of apoptotic cells- A review. Indian J Cancer. 2013; 50:274-283. [Crossref]
- Yasuhara S, Zhu Y, Matsui T, Tipirneni N, Yasuhara Y, Kaneki M, Rosenzweig A, Martyn JA. Comparison of comet assay, electron microscopy, and flow cytometry for detection of apoptosis. J Histochem Cytochem. 2003; 51:873-885. [Crossref]
- Roos WP, Kaina, B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013; 332:237-248. [Crossref]
- Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prévost MC, Brothers G, Mak TW, Penninger J, Earnshaw WC, Kroemer G. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000; 192:571-580. [Crossref]
- Baeshen NA, Elkady AI, Abuzinadah OA, Mutwakil MH. Potential anticancer activity of the medicinal herb, Rhazya stricta, against human breast cancer. Afr J Biotechnol. 2012; 11:8960-8972. [Crossref]
- Cipriani B, Borsellino G, Knowles H, Tramonti D, Cavaliere F, Bernardi G, Battistini L, Brosnan CF. Curcumin inhibits activation of Vγ9Vδ2 T cells by phosphoantigens and induces apoptosis involving apoptosis-inducing factor and large scale DNA fragmentation. J Immunol. 2001; 167:3454-3462. [Crossref]
- Zhou H, Xu M, Gao Y, Deng Z, Cao H, Zhang W, Wang Q, Zhang B, Song G, Zhan Y, Hu T. Matrine induces caspase-independent programmed cell death in hepatocellular carcinoma through bid-mediated nuclear translocation of apoptosis inducing factor. Mol Cancer. 2014; 13:59. DOI: 10.1186/1476-4598-13-59. [Crossref]
- Esmaeili-Mahani S, Falahi F, Yaghoobi MM. Proapoptotic and antiproliferative effects of Thymus caramanicus on human breast cancer cell line (MCF-7) and its interaction with anticancer drug vincristine. Evid Based Complement Alternat Med. 2014; 2014:893247. DOI: 10.1155/2014/893247. [Crossref]
- Molinari M. Cell cycle checkpoints and their inactivation in human cancer. Cell Prolif. 2000; 33:261–274. [Crossref]
- Benada J, Macurek L. Targeting the checkpoint to kill cancer cells. Biomolecules. 2015; 5:1912-1937. [Crossref]
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009; 137:413-431. [Crossref]
- Selivanova G. Wild type p53 reactivation: From lab bench to clinic. FEBS Lett. 2014; 588:2628-2638. [Crossref]
- Chi SW. Structural insights into the transcription-independent apoptotic pathway of p53. BMB Rep. 2014; 47:167-172. [Crossref]
- Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene. 2008; 27:6507-6521. [Crossref]
- Stivala LA, Cazzalini O, Prosperi E. The cyclin-dependent kinase inhibitor p21CDKN1A as a target of anti-cancer drugs. Curr Cancer Drug Targets. 2012; 12:85–96. [Crossref]
- Caldon CE, Daly RJ, Sutherland RL, Musgrove EA. Cell cycle control in breast cancer cells. J Cell Biochem. 2006; 97:261-274. [Crossref]
- Roy PG, Thompson AM. Cyclin D1 and breast cancer. Breast. 2006; 15:718-727.
- Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011; 11:558-572. [Crossref]
- Meeran SM, Katiyar SK. Cell cycle control as a basis for cancer chemoprevention through dietary agents. Front Biosci. 2008; 13:2191-2202. [Crossref]