Phage display technology is a powerful tool for the selection of peptides or proteins that specifically bind a target of interest. Antibody fragment selection is one of the most successful applications of this technique. Although modern simple gene manipulation techniques have emerged and the demand for phage display technology has increased, very few phage display vectors are readily available. Therefore, the construction of phage display vectors, which are prepared using common reagents and simple procedures, was attempted. The fragments from the pUC118 vector, the M13KO7 genome and short synthetic DNA were assembled and modified, and the three phage display vectors were constructed. When the human cellular retinoic acid binding protein II (hCRABPII) was cloned into a pdvec2 vector, protein display was successfully observed. Simple gene structure and the use of easily-accessible components will facilitate applications of the phage display system.
gene III, hCRABPII, M13KO7, phage display, pUC118
Phage display system is one of the most widely used modern biotechnological methodologies [1]. Since the first protein display on phage was reported [2], it has been used in the discovery of many functional proteins, especially substrate binding proteins. The gene III product of filamentous bacteriophage is the most utilized phage surface protein for displaying foreign peptides or proteins as it can accept a wide range of molecular mass without denaturation.
Screening peptides that bind to specific targets is one of the main applications of the phage display system. Mapping epitopes and mimotopes, identifying peptide ligands, and mapping post-translational modification substrate sites are other commonly performed applications [1]. Additionally, new applications such as screening cell-surface binding peptides or drug delivery carrier peptides are being developed.
Another notable application of the phage display system is an antibody-display library [3]. The antibody domains, such as single-chain variable fragment (scFv), can be displayed on the phage particle. An scFv that binds to a disease-related factor can be engineered into a full-body immunoglobulin molecule and developed into a therapeutic antibody. A number of antibodies that have been developed with the phage display system are approved for therapy.
In the early days of phage display technology, gene manipulation was rather complicated and there were many technological challenges. Therefore, only a limited number of laboratories with the technological knowledge were able to perform phage display experiments. However, with the accumulation of technical experience, the demand for a convenient phage display vector and protocols to construct quality libraries have increased. Furthermore, the wide use of genetic manipulation techniques in synthetic biology has made the development of phage display system libraries less complex. A simple, functional, and easily available phage display vector will support the expanding applications in many laboratories. Therefore, we attempted to develop a functional phage display vector using modern, simple laboratory procedures and common components. The rearrangement of the polynucleotide components was carried out using simple gene synthesis protocols. The display of human retinoic acid binding protein was successfully confirmed using the newly engineered vector.
Materials
Reagents were purchased from Wako Pure Chemicals Industries Ltd. (Osaka, Japan) or Sigma-Aldrich (Tokyo, Japan). Oligonucleotides were purchased from Eurofins Genomics (Tokyo, Japan; sequences in Table 1). DNA polymerases, restriction enzymes, and modification enzymes were purchased from Takara Biotech (Shiga, Japan), Toyobo (Osaka, Japan), or New England Biolabs (Tokyo, Japan). The standard plasmid vector pUC118 [4] was obtained from a laboratory stock. The helper phage M13KO7 was obtained from New England Biolabs.2.2. Construction of pdvec0 plasmid
Table 1. List of oligonucleotides and their sequences.
Oligonucleotide |
Sequence (5′ to 3′) |
pUC118_R |
AGCTGTTTCCTGTGTGAAATTGTTATCCGC |
pUC118_F |
ATGACCATGATTACGAATTCGAGCTCGGTA |
g3N1_F |
AGGGGCCTCTGCAGAAACTGTTGAAAGTTG |
g3N3_R |
GAATTCAAGCTTAAGACTCCTTATTACGCA |
g3sig_mcs_F1 |
ATGAAAAAATTATTATTCGCAATTCCTTTA |
g3sig_mcs_F2 |
GTTGTTCCTTTCTATTCTCACTCGGCCGGC |
g3sig_mcs_F3 |
TCGGCCCATATGTAATGATAGTAATGATAG |
g3sig_mcs_F4 |
CTCGAGGAATTCGGGGCCTCAGGGGCCGCG |
g3sig_mcs_R1 |
ATAGAAAGGAACAACTAAAGGAATTGCGAA |
g3sig_mcs_R2 |
TTACATATGGGCCGAGCCGGCCGAGTGAGA |
g3sig_mcs_R3 |
CCCGAATTCCTCGAGCTATCATTACTATCA |
g3sig_mcs_R4 |
TTCAACAGTTTCTGCAGAGGCCCCTGAGGC |
PD_N_S |
GACTACAAAGATGATGATGATAAGGGCTCGGCCCATATGTAA |
PC_Nde_A |
CACAATCAATAGAAAATTCGTACGGTTTACCAGCGCTAAAGAC |
PC_Nde_S |
GTCTTTAGCGCTGGTAAACCGTACGAATTTTCTATTGATTGTG |
PD_N_A |
CTTATCATCATCATCTTTGTAGTCCGAGTGAGAATAGAAAGG |
pdvec2_Fv |
GGCCTCTGCAGAAACTGTTGAAAGTTG |
pdvec2_Rv |
TATTTCATAGCTGTTTCCTGTGTGAAATTG |
pdvec2_F0 |
CAATTTCACACAGGAAACAGCTATGAAATA |
pdvec2_F1 |
CCTGCTGCCGACCGCTGCTGCTGGTCTGCT |
pdvec2_F2 |
GCTCCTCGCTGCCCAGCCGGCGATGGCCAT |
pdvec2_F3 |
GGACTACAAAGATGATGATGATAAGGCCGG |
pdvec2_F4 |
CTCGGCCCATATGTAATGATAGTAATGATA |
pdvec2_F5 |
GCTCGAGGAATTCGGTCACCACCACCACCA |
pdvec2_F6 |
CCACTAGGGGGCCTCAGGGGCCTCTGCAGA |
pdvec2_R0 |
GCGGTCGGCAGCAGGTATTTCATAGCTGTT |
pdvec2_R1 |
TGGGCAGCGAGGAGCAGCAGACCAGCAGCA |
pdvec2_R2 |
TCATCTTTGTAGTCCATGGCCATCGCCGGC |
pdvec2_R3 |
TACATATGGGCCGAGCCGGCCTTATCATCA |
pdvec2_R4 |
CCGAATTCCTCGAGCTATCATTACTATCAT |
pdvec2_R5 |
GAGGCCCCCTAGTGGTGGTGGTGGTGGTGA |
pdvec2_R6 |
CAACTTTCAACAGTTTCTGCAGAGGCCCCT |
pdvec2_Fi |
CAATTTCACACAGGAAACAGCTATGAAATA |
pdvec2_Ri |
CAACTTTCAACAGTTTCTGCAGAGGCC |
HCRABP_F01 |
AGACTACCCCTCTAGAAATAATTTTGTTTA |
HCRABP_F02 |
GGAGATATACATATGCCGAATTTCTCTGGC |
HCRABP_F03 |
TCATTCGTTCTGAAAACTTTGAAGAACTGC |
HCRABP_F04 |
GGGCGTCAACGTAATGCTGCGTAAAATCGC |
HCRABP_F05 |
GCGAGCAAGCCGGCGGTTGAAATCAAACAG |
HCRABP_F06 |
CCTTCTATATCAAAACCTCCACGACCGTAC |
HCRABP_F07 |
AATCAACTTCAAAGTTGGTGAGGAGTTCGA |
HCRABP_F08 |
GTAGACGGTCGTCCATGCAAGTCCCTGGTT |
HCRABP_F09 |
CTGAAAACAAGATGGTTTGTGAACAGAAAC |
HCRABP_F10 |
CGAAGGCCCGAAAACCTCCTGGACCCGTGA |
HCRABP_F11 |
GATGGCGAACTGATCCTGACGATGACCGCG |
HCRABP_F12 |
TATGCACGCGCGTTTACGTGCGTGAAGGCG |
HCRABP_R01 |
GTATATCTCCTTCTTAAAGTTAAACAAAAT |
HCRABP_R02 |
GAACGAATGATTTTCCAGTTGCCAGAGAAA |
HCRABP_R03 |
GTTGACGCCCAGAACCTTCAGCAGTTCTTC |
HCRABP_R04 |
GCTTGCTCGCAGCCGCAACTGCGATTTTAC |
HCRABP_R05 |
ATATAGAAGGTGTCGCCTTCCTGTTTGATT |
HCRABP_R06 |
GAAGTTGATTTCGGTGGTGCGTACGGTCGT |
HCRABP_R07 |
GACCGTCTACGGTCTGTTCCTCGAACTCCT |
HCRABP_R08 |
TTGTTTTCAGATTCCCATTTAACCAGGGAC |
HCRABP_R09 |
CGGGCCTTCGCCTTTCAGCAGTTTCTGTTC |
HCRABP_R10 |
GTTCGCCATCATTAGTCAGTTCACGGGTCC |
HCRABP_R11 |
CGCGTGCATACTACGTCATCCGCGGTCATC |
HCRABP_R12 |
CACCCTGAATTCCTCGAGGCCGCCTTCACG |
All genetic manipulations were performed according to standard procedures [5], unless otherwise noted. The phage display vector, pdvec0, was constructed as follows. The vector fragment was prepared using the pUC118 vector as a template. The oligonucleotides pUC118_R and pUC118_F were phosphorylated with T4 polynucleotide kinase (Toyobo) according to the manufacturer's instructions. Polymerase chain reaction (PCR) was performed using the phosphorylated pUC118_R and pUC118_F primers, the pUC118 vector, and KOD Plus Neo DNA polymerase (Toyobo) according to the manufacturer's instructions. The samples were purified from the reaction mix using a Wizard SV Gel and PCR Clean-Up System (Promega, Tokyo, Japan). The gene III fragment of filamentous phage was prepared using the M13KO7 bacteriophage genome as a template. The genome was isolated from M13KO7 phage suspension by phenol/chloroform extraction and ethanol precipitation. PCR was performed using g3N1-F and g3N1-R primers, the M13KO7 genome, and KOD Plus Neo DNA polymerase. The PCR product was purified as described above. The short DNA fragment was prepared using a modified two-step DNA strand assembly technique [6]. First, a thermal cycler reaction of 94 °C for 2 min, followed by 30 cycles of 98 °C for 10 s, 55 °C for 30 s, and 68 °C for 30 s were performed with 2.5 pmol each of g3sig_mcs_F1 to F4, R1 to R4, and 1 U KOD Plus Neo polymerase. PCR mix (50 μL) containing 1 μL of the thermal cycler reaction, phosphorylated g3sig_mcs_F1 and g3sig_mcs_R1 primers, and 1 U KOD Plus Neo polymerase was prepared. PCR for the assembled fragment amplification was performed at 94 °C for 2 min, followed by 30 cycles of 98°C for 10 s and 68°C for 30 s. The plasmid was purified as described above. The fragment from pUC118 was digested with HindIII, and the fragments of gene III and synthetic DNA were digested with PstI. The three fragments were ligated and E. coli JM109 was transformed using the purified ligation mixture. DNA insertion was confirmed by plasmid sequencing.
Construction of pdvec1 plasmid
The vector plasmid, pdvec1, was constructed as follows. PCR was performed using the pdvec0 vector as a template. The insert fragment was obtained from the reaction mixture containing PD_N_S and PC_Nde_A primers. The vector fragment was obtained from the reaction mixture containing PC_Nde_S and PD_N_A primers. The purified fragments were assembled using the circular polymerase extension cloning (CPEC) procedure [7], and the pdvec1 vector was generated. The nucleotide sequence was confirmed by sequencing.
Construction of pdvec2 plasmid
The plasmid vector with pelB leader sequence at the upstream region of the cloning site was constructed as follows. The vector fragment was amplified by PCR using pdvec2_Fv and pdvec2_Rv primers, and pdvec1 as a template. The assembled fragment was constructed using the two-step DNA strand assembly technique. The mixture for the first PCR contained 2.5 pmol oligonucleotides (pdvec2_F0 to F6 and pdvec2_R0 to R6). The second PCR for assembled fragment amplification was performed using pdvec2_Fi and pdvec2_Ri primers. Then, the vector and the insert fragments were assembled by CPEC reaction. The nucleotide sequence was confirmed by sequencing.
Cloning of human retinoic acid binding protein to pdvec1 and pdvec2
The amino acid sequence of the human cellular retinoic acid binding protein II (hCRABPII) was obtained from ID 2FR3 [8] in the Protein Data Bank [9] (www.wwpdb.org). The structural gene sequence that codes the hCRABPII amino acid sequence was designed using the Gene Designer software [10]. Codon usage and gene structure were biased for high expression in E. coli. From the designed nucleotide sequence, 24 oligonucleotides (HCRABP_F01 to F12 and HCRABP_R01 to R12) were synthesized. The oligonucleotides are assembled using the two-step DNA strand assembly technique. After PCR amplification of assembled gene fragment with HCRABP_F01 and HCRABP_R01, a 490-bp fragment was digested with NdeI and XhoI restriction enzymes. The fragment was then ligated to each of pdvec1 and pdvec2 vectors that were digested with the same restriction enzymes. The nucleotide sequence around the insert was confirmed by sequencing for each vector.
Phage amplification
Display phage was prepared as follows [1]. Escherichia coli TG1, harboring the above-constructed phagemid, was inoculated in 10 mL 2×YT medium with 2% (w/v) glucose and 100 μg/mL ampicillin, and cultured at 30°C. When the culture OD600 reached 0.7 to 1.0, M13KO7 was added at a multiplicity of infection (MOI) 5. After 30 min incubation without shaking, the culture was centrifuged at 3000 ×g for 5 min, and E. coli was washed with fresh 2×YT medium containing 100 μg/mL ampicillin and 50 μg/mL kanamycin (2×YTAK). After another wash, E. coli was resuspended in 10 mL 2×YTAK medium and cultured overnight. Subsequently, the culture was centrifuged, and the supernatant was filtered through a 0.2 μm syringe filter (DISMIC-25cs, Advantec). One-fifth volume of 20% (w/v) polyethylene glycol and 2.5 M NaCl solution was added to the filtrate, incubated on ice for approximately 1 h, and centrifuged. The precipitate was resuspended in phosphate buffered saline (PBS) with 50% (v/v) glycerol and stored at -30 °C or -80 °C until use. Phage titration was carried out using the mid-log phase E. coli JM109 culture as a carrier, and the titer was calculated from the colony number on LB solid media containing 100 μg/mL ampicillin.
Phage enzyme linked immunosorbent assay (ELISA)
Standard phage sandwich ELISA was performed as follows. Monoclonal ANTI-FLAG M2 antibody (Sigma) was diluted in PBS, and 100 μL aliquot of the solution, containing 1 μg antibody, was dispensed to a microtiter plate (Immuno Module F8, Nunc). After incubation at 4°C overnight, each well was washed three times with PBS containing 0.05% (v/v) Tween 20 (PBS-T). Then, 200 μL of 3% (w/v) bovine serum albumin (BSA; Fraction V, Sigma) in PBS was dispensed and incubated for 1 h at 37 °C for blocking. After blocking, each well was washed three times with PBS-T, and the phage solution diluted in PBS with 1% (w/v) of skimmed milk (MPBS) was added. The strip was incubated for 1 h at 37°C and washed three times with PBS-T. MPBS (100 μL) containing peroxidase-conjugated anti-M13, Fd, F1 filamentous phages (PROGEN Biotechnik, Germany) was added to each well, incubated for 1 h at 37°C, and washed three times with PBS-T. Then, 100 μL of 3,3′,5,5′-tetramethylbenzidine (TMB) solution (1-Step Ultra TMB-ELISA Substrate Solution, Thermo Fisher Scientific) was dispensed to each well and incubated for 30 min in the dark. The chromogenic reaction was stopped by addition of 100 μL of 1 M H2SO4, and the absorbance was measured at 450 nm on a microtiter plate reader (Viento XS, DS Pharma Biomedical, Japan).
Construction of the vectors for phage display
Although phage display systems have become more readily accessible and more applications of phage display technology are being developed, the number of phage display vectors available in the market is limited. Additionally, as display vectors need to be optimized for the protein or peptide of interest a vector with a simple genetic structure is desirable. This warrants the engineering of phage display vectors using readily-available components. The schematic diagram of vector construction is shown in Figure 1.
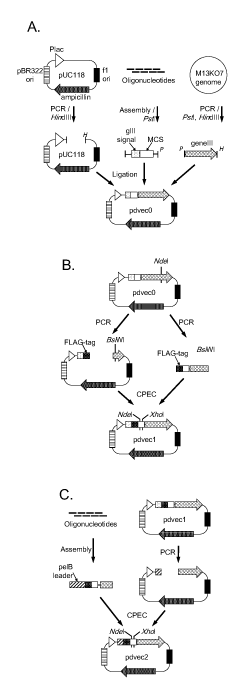
Figure 1. Schematic diagram of phage display vectors construction. (A) pdvec0 construction. A vector fragment of pUC118 was amplified by polymerase chain reaction (PCR) and digested with HindIII (H). Assembled oligonucleotides, containing gene III signal and multicloning site (MCS), were digested with PstI (P). The PCR-amplified gene III fragment was digested with PstI and HindIII. These three fragments were ligated to generate the pdvec0 vector. (B) pdvec1 construction. During the PCR of vector fragment, a FLAG-tag coding sequence was generated downstream of the gene III signal, and the NdeI restriction site in gene III was synonymously replaced by BsiWI restriction site. The margin sequences of FLAG-tag and gene III partial sequences were also generated at the terminals of the insert fragment by PCR. These two fragments were assembled by CPEC reaction. (C) pdvec2 construction. The assembled fragment contained a pelB leader sequence instead of a gene III signal sequence. The fragment was ligated with the digested vector by CPEC reaction. Both pdvec1 and pdvec2 have unique NdeI and XhoI restriction sites in the MCS.
The first phagemid vector, pdvec0, was constructed using three DNA fragments. The pUC118 plasmid and the M13KO7 helper phage are common reagents in many laboratories. The first and the second fragments were derived from pUC118 and M13KO7 genome, respectively, using PCR. The third fragment was constructed using eight short oligonucleotides to generate the gene III signal peptide coding sequence and multicloning site. Two-step assembly of the plasmid was achieved.
The second vector, pdvec1, was a derivative of pdvec0. An NdeI restriction site in gene III was replaced with a BsiWI restriction site because the multicloning site carries a second NdeI restriction site. A FLAG-tag coding sequence was inserted between the signal coding sequence and multicloning site for detection of displayed protein by ELISA.
The third vector, pdvec2, was constructed by replacement of the upstream region of gene III of pdvec1 with a synthetic fragment. The signal sequence was replaced with the pelB leader sequence as the display efficiency was found to vary by the donor protein or peptide, as described below. Additionally, more restriction sites, a polyhistidine-tag coding sequence, and an amber stop codon (TAG) were generated in the multicloning site. Fragment assembly and CPEC were achieved.
Display of hCRABP II
hCRABP II is associated with cell differentiation and development and is the mediator of retinoic acid in the cell [8,11]. HCRABPII preferably binds to all-trans retinoic acid with high affinity [12], through its ligand binding site [11].
The design and development of small molecules that bind to specific proteins are valuable. Such small molecules can be developed into potential drugs and may be used in biochemical analysis or diagnostics. We attempted to display the hCRABPII on phage particles using engineered phagemid vectors, as a screening tool for small molecules binding to hCRABP.
The synthesized hCRABPII-coding DNA fragment was introduced into pdvec1 and pdvec2 vectors. The display phage was prepared and the level of the display was measured by ELISA (Figure 2). Although very small signals were observed even at high phage titer (~3 x 1012 cfu) in the pdvec1-based clone, a large signal accompanied with input phage titer was observed for pdvec2-based construct. Differences in the signal sequence between the gene III signal (pdvec1) and pelB leader (pdvec2) might affect display efficiency. However, when a single chain variable region library of human immunoglobulin was cloned into pdvec1 and pdvec2, higher display efficiency of pdvec1 was observed (data not shown). The influence of signal sequence on display efficiency and the relation of the type of the signal sequence with the target protein has been widely reported [13]. Thus, the screening of the suitable type of signal sequence may be required for the initial experimental attempt of display for each specific target protein or peptide. In the gene III sequence confirmation process by sequencing, the three-nucleotide replacement was found against the sequence from New England Biolabs Website (https://international.neb.com/-/media/nebus/page-images/tools-and-resources/interactive-tools/dna-sequences-and-maps/text-documents/m13ko7gbk.txt?la=en). These mutations did not affect hCRABPII display efficiency and were presumably from the high mutation rate of the bacteriophage genome. However, it might be required to check gene III mutations when a foreign protein fails to be displayed by similar self-made display vectors.
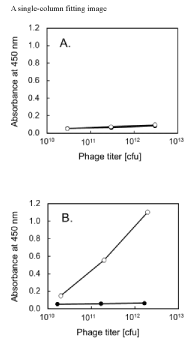
Figure 2. Human cellular retinoic acid binding protein II (hCRABPII) display phage validation by enzyme linked immunosorbent assay (ELISA). ELISA signal of absorbance at 450 nm for the input phage titer is shown for vectors (A) pdvec1, (B) pdvec2. The display phages were prepared using the vector harboring hCRABPII (open circle), or the unmodified vector (closed circle).
The efficiency of library construction using these vectors needs to be verified. Our current trials of mutant library construction using pdvec2 and synthetic gene fragments of foreign protein mutants using the CPEC procedure has been mostly successful (data not shown). We are currently evaluating the competency of these vectors for displaying multiple proteins/peptides. Furthermore, we are currently verifying successful selection of active mutants from several constructed mutant libraries, which is another point of interest. Our results from these experiments will be reported in peer-reviewed journals.
Three types of vectors for phage display were successfully constructed. We used readily-available reagents, and fragment synthesis and component assembly were successfully performed using simple and up-to-date genetic manipulation techniques. Incorporating ligation-free assembly techniques, such as the Gibson assembly [14] or In-Fusion (Clontech), would further simplify vector synthesis and library construction. This simplified process will facilitate foreign protein display, improvement of display efficiency, library generation, and successful clonal screening. Currently, we are generating a library for a wide variety of proteins including hCRABPII using the pdvec2 vector.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
The author(s) declare that they have no competing interests.
- Kay BK, Winter J, McCafferty J, et al. (1996) Phage display of peptides and proteins?: a laboratory manual. Academic Press.
- Smith GP (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228, 1315–1317. [Crossref]
- Frenzel A, Schirrmann T, Hust M (2016) Phage display-derived human antibodies in clinical development and therapy. MAbs 8, 1177–1194. [Crossref]
- Vieira J, Messing J (1987) Production of single-stranded plasmid DNA. Methods Enzymol 153: 3-11. [Crossref]
- Green MR, Sambrook J (2012) Molecular cloning?: a laboratory manual. Cold Spring Harbor Laboratory Press.
- Xiong AS, Yao QH, Peng RH, Li X, Fan HQ, et al. (2004) A simple, rapid, high-fidelity and cost-effective PCR-based two-step DNA synthesis method for long gene sequences. Nucleic Acids Res 32: e98. [Crossref]
- Quan J, Tian J (2009) Circular polymerase extension cloning of complex gene libraries and pathways. PLoS One 4: e6441. [Crossref]
- Vaezeslami S, Mathes E, Vasileiou C, Borhan B, Geiger JH (2006) The Structure of Apo-wild-type Cellular Retinoic Acid Binding Protein II at 1.4 Å and its Relationship to Ligand Binding and Nuclear Translocation. J Mol Biol 363, 687–701. [Crossref]
- Berman H, Henrick K, Nakamura H (2003) Announcing the worldwide Protein Data Bank. Nat Struct Biol 10: 980. [Crossref]
- Villalobos A, Ness JE, Gustafsson C, Minshull J, Govindarajan S (2006) Gene Designer: a synthetic biology tool for constructing artificial DNA segments. BMC Bioinformatics 7: 285. [Crossref]
- Norris AW, Rong D, d'Avignon DA, Rosenberger M, Tasaki K, et al. (1995) Nuclear magnetic resonance studies demonstrate differences in the interaction of retinoic acid with two highly homologous cellular retinoic acid binding proteins. Biochemistry 34, 15564–15573. [Crossref]
- Redfern CP, Wilson KE (1993) Ligand binding properties of human cellular retinoic acid binding protein II expressed in E. coli as a glutathione- S -transferase fusion protein. FEBS Lett. 321, 163–168. [Crossref]
- Paschke M (2006) Phage display systems and their applications. Appl Microbiol Biotechnol 70: 2-11. [Crossref]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, et al. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343-345. [Crossref]