Mixed lineage leukemia (MLL) is an aggressive blood cancer that results from genetic alterations in the MLL1 gene. This gene encodes an enzyme that methylates histone H3 on lysine 4 (H3K4) as a part of the MLL1 multi-protein complex. MLL-related genetic alterations create fusion proteins that render MLL1 incapable of its methyltransferase activity, with particularly devastating effects at homeobox genes. Problematically, higher eukaryotes contain many functionally redundant complexes that complicate the study of MLL1 and associated cancers in a living system. Moreover, the translocations that lead to the formation of MLL1 fusion proteins are variable, generating similarly variable fusion proteins. This inconsistency further complicates the use of MLL1 as a drug target. However, several accessory proteins within the complex are required for catalytic activity, and present possible drug targets themselves. Herein we present an in vivo system for the study of the MLL1 complex in Saccharomyces cerevisiae, making use of the homologous Set1/COMPASS complex. We genetically replaced COMPASS members from S. cerevisiae with their human homologs using antibiotic resistance cassettes, and subsequently performed phenotypic characterization of chimeric COMPASS/MLL1 complexes, assessing global H3K4 methylation status. Selected chimeric yeast-human methyltransferase complexes conferred catalytic activity at varying degrees, while others did not confer methyltransferase activity. Notably, we observed H3K4 dimethylation levels comparable to wild type when human Ash2L replaced yeast Bre2 but reduced levels of H3K4 trimethylation with this same chimeric complex. Together, these data represent a proof of concept for simplifying the study of this clinically important protein complex in a tractable in vivo system, and also offer mechanistic insight into the functional role of a catalytically essential accessory protein within the MLL1 complex through our model.
histone H3, methyltransferase, Set1, COMPASS, epigenetics
Genome rearrangements and the MLL1 complex
Drastic genomic rearrangements including chromothripsis, aneuploidies, and translocations are invariably detrimental to all human cells. Faulty regulation of gene expression following such an event often contributes to disease progression, perhaps most notably in cancer. In the case of mixed lineage leukemia (MLL), a particularly deadly form of cancer, translocations involving the histone methyltransferase gene MLL1 are known to alter gene expression in such a way that normal hematopoiesis is disrupted, leading to leukemogenesis [1,2]. The MLL1 gene encodes the catalytic subunit of a multi-protein complex (termed the MLL1 complex) that performs crucial histone H3 lysine 4 (H3K4) methylation, a modification usually associated with transcriptional activation [3]. Via this epigenetic mechanism, the MLL1 complex exerts regulatory control over many genes, including homeobox (HOX) genes, which are critically important to normal hematopoiesis. In the disease state following a translocation, MLL1 forms fusion proteins with at least 70 partner proteins that alter the MLL1 complex’s catalytic activity, and thereby affect the expression of HOX genes, disrupt normal hematopoiesis, and trigger leukemogenesis [4,5]. MLL1 fusion proteins have also been shown to deleteriously alter p53-mediated response to DNA damage [6].
Therapeutic potential of epigenetic modifiers
Despite the unfortunately poor prognosis of affected individuals, the mechanistic basis of mixed lineage leukemia warrants cautious optimism for the long-term development of targeted therapies. It is generally accepted that epigenetic modifiers represent feasible targets for therapeutic intervention, and other such proteins have seen some success as ‘druggable’ targets for cancer treatment as evidenced by the FDA-approved histone deacetylase inhibitor vorinostat used for the treatment of cutaneous T-cell lymphoma [7]. Problematically, MLL1 fusion proteins are structurally inconsistent from one patient to another, making MLL1 itself an impractical drug target. Some of the accessory proteins within the MLL1 complex, however, are required for catalytic activity, affect the specificity and catalytic activity of the complex [8,9], and are post-translationally modified [10-12]. Together, these factors suggest that the accessory proteins of the MLL1 complex play an important role in the regulation of epigenetic modification by MLL1. Based on this regulatory role, these proteins may represent viable targets for therapeutic intervention. Such studies have already yielded some clinical progress and researchers are developing new and potent inhibitors of MLL1 complex member interaction (specifically between MLL and WDR5) with the eventual goal of use in cancer treatment [13-16].
Results and limitations of prior in vitro studies
Although numerous studies have been published that attempt to elucidate the mechanism of action of the core MLL1 complex in relation to leukemia and H3K4 trimethylation, all such studies have been limited to in vitro models [11,17-19]. Recent in vitro studies were all found that the methyltransferase subunit necessary for the trimethylation of H3K4 is activated by a heterodimer of Ash2L and RbBP5 (two accessory proteins in the MLL1 protein complex) in a two-step process that may potentially be conserved across all SET-containing histone methyltransferases. However, that protein interactions and the degree of their necessity for complex function remain unclear [17]. The extent to which each member regulates and participates in the actions of the methyltransferase are, however, still unclear. In vivo study of the MLL1 protein complex is extremely difficult due to the presence of functionally redundant H3K4 methyltransferase complexes present in human cells such as MLL 2-4, Set1d1A and Set1d1B [20]. These complexes share many of the same accessory proteins; therefore, differentiating between the specific regulatory roles of the accessory proteins on catalytic activity in mammals is challenging, if not impossible [21]. Consequently, most research on these intra-complex interactions has come from in vitro models utilizing recombinant proteins or computer modeling [17,18,22,23]. Several structural studies have been performed yielding insight into conformational changes of MLL as well [12,17,24]. While these studies have provided key insights into the MLL1 complex, exhaustive functional characterization of the accessory proteins is not possible in silico, in vitro, or in bacteria because these proteins are endogenously post-translationally modified. These modifications, in effect, contribute to complex activity and/or specificity. Thus, in vitro studies ignore the interconnectedness of the complexes due to (relatively unknown) endogenous mechanisms and therefore may reach incorrect conclusions from only partial data. To faithfully capture MLL1 complex function in a biologically relevant context, an in vivo model is necessary. Saccharomyces cerevisiae (S. cerevisiae) provides a tractable in vivo model with which to study mis regulation of H3K4 methylation by the MLL complex.
An in vivo model for MLL1 complex function in S. cerevisiae
Previous studies on MLL1 have not been performed in vivo due to the difficulty of performing the experiment in metazoan cells. Importantly, posttranslational modifications have been identified on MLL1 complex members that occur only in the context of a eukaryotic cell [10,11,12,24]. As previously mentioned, MLL1 has numerous functionally similar proteins in higher eukaryotes and therefore its isolated study is challenging. However, understanding and analyzing the posttranslational modifications that have been identified on MLL1 complex members are essential to holistically and realistically study the protein complex function.
It has been shown that the SET domain of the MLL1 protein methylate’s itself and Ash2L, and this only occurs in in the absence of histone H3, whereas unmethylated H3 serves as a stoichiometric inhibitor of auto-methylation [11]. MLL1 auto-methylation appears more prevalent, however, in the absence of the MLL1 core complex members than in their presence [11]. Such modifications likely affect intra-complex interactions that alter catalytic function in vivo, and the effects of these modifications represent key gaps in present knowledge of the MLL1 complex. Additionally, outside of a eukaryotic context, intra-complex regulation dependent on other histone modifications cannot be evaluated. These histone trans-regulatory networks have already been shown as modifying factors for the MLL1 complex [8]. In our study, we sought to study the accessory proteins of the human MLL1 complex in a eukaryotic system that lacks the problematic functionally redundant MLL1 homologs present in metazoans. The commonly used model organism S. cerevisiae was ideal for this purpose since it is both eukaryotic and contains only a single homolog to the MLL1 complex. The homologous yeast protein complex is termed COMPASS (Complex of Proteins Associated with Set1) [25]. Research centering on COMPASS has begun to define the roles of the accessory proteins associated with yeast Set1, the homolog of human MLL1. Individual COMPASS subunits (Set1, Bre2, Swd1, and Swd3) have been shown to confer proteomic stability and integrity, as well as regulate H3K4 methylation both globally and specifically [3,26,27].
In this study, we have adapted the tools available in yeast genetics to the biochemical study of the clinically important MLL1 complex. We show that specific COMPASS accessory proteins can be replaced by their human homolog and yield varying degrees of global H3K4 methylation. These interspecies chimeric H3K4 methyltransferase complexes have varying functionality based on the specific complex proteins present. Most notably, we show that H3K4 dimethylation levels were comparable to wild type when human Ash2L replaced yeast Bre2. However, this same chimeric complex showed reduced levels of H3K4 trimethylation compared to wild type. This data represents a proof of concept, demonstrating that the study of the MLL1 complex can be studied in a tractable in vivo model. Moreover, this data also provides mechanistic insight into a functionally important accessory protein within the human complex.
Yeast growth assays
Serial dilution yeast growth assays for the indicated strains constructed in this study were performed as previously described [28].
Yeast strains, strain construction and growth media
All S. cerevisiae strains used in this study were W303 and BY4741, both derived from strain S288C (Table 1). All yeast procedures were performed at 30°C and followed as stated in Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual [29]. The genes encoding the four proteins of interest in COMPASS were deleted by a method of homologous recombination wherein an antibiotic resistance cassette is inserted into the gene of interest via transformation (Table 2). Successful transformations were assessed through genotyping with polymerase chain reaction (PCR) and gel electrophoresis.
Table 1. Saccharomyces cerevisiae strains used in the course of this study
Yeast Strain |
Strain Name |
W303: MATα, leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15, set1::KanMX |
set1∆ |
W303: MATα, leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15, swd1::KanMX |
swd1∆ |
W303: MATα, leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15, bre2::KanMX |
bre2∆ |
W303: MATα, leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15, swd3::KanMX |
swd3∆ |
W303: MATα, leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15, set1::KanMX, bre2::HphMX |
set1∆bre2∆ |
W303: MATα, leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15, set1::KanMX, bre2::NatMX, swd3::HphMX |
set1∆bre2∆swd∆ |
BY4741: MATα, leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15, set1::KanMX, bre2::HphMX, swd1::NatMX |
set1∆bre2∆swd1∆ |
Table 2. Yeast gene replacements
Yeast Gene |
Deletion antibiotic resistance cassette |
Human Gene |
Expression Plasmid |
Set1 |
kanr |
MLL |
pAG413/His |
Bre 2 |
patr |
Ash2L |
pAG415/Leu |
Swd1 |
natr |
RbBP5 |
pAG414/Trp |
Swd3 |
hphr |
WDR5 |
pAg416/Ura |
Gene replacements were performed with one or more of the following resistance genes: kanMX (pUG6), natMX4 (pAG25) or hphMX4 (pAG32) from Euroscarf and then confirmed by PCR genotyping as described previously [30,31,32]. Primers specific for each gene deletion used to amplify the antibiotic resistance cassettes are listed in Table S1. All deletion strains were genotyped by PCR using a primer specific to the 5’ or 3’ end of the coding region along with a primer specific to the antibiotic cassette insert (“genotyping primers” listed in Table S1). Drug containing plates were prepared using the following concentrations: 200μg/mL of Geneticin (Invitrogen), 100μg/mL Nourseothricin sulfate (SIGMA) or 300μg/mL Hygromycin B (SIGMA). Yeast were propagated according to standard procedures either in rich media (YPD), YP media containing 20% galactose or in appropriate selective media.
Table S1. Oligonucleotide Primers
Primer Description |
Primer sequence |
set1 5’ disruption primer |
TATTTGTTGAATCTTTATAAGAGGTCTCTGCGTTTAGAGACAGCTGAAGCTTCGTACGC |
set1 3’ disruption primer |
TGTTAAATCAGGAAGCTCCAAACAAATCAATGTATCATCGGCATAGGCCACTAGTGGATCTG |
set1 5’ genotyping primer |
GGTTTTAATGGTATTGTTTTGG |
set1 3’ genotyping primer |
CAATTTCGTGTGTCAATTGAAC |
bre2 5’ disruption primer |
GCCATAATTGGACGAAGACAAATAATTCACTTCCTTAATACAGCTGAAGCTTCGTACGC |
bre2 3’ disruption primer |
ACTTTCAGTGTGTTTTAATTATTCTTCTTTGAATGCTGCTGCATAGGCCACTAGTGGATCTG |
bre2 5’ genotyping primer |
CAAAGAAACACATGGTACCTC |
bre2 3’ genotyping primer |
CTATTTCTCCAGATAACTACATG |
swd1 5’ disruption primer |
AGTCCTTTTAGTTTGTGTATATCAGCTGGTTCTTTTCGTTCAGCTGAAGCTTCGTACGC |
swd1 3’ disruption primer |
GTCAGTTGCCTATTTAAACTTTCTCCTTTTGAAGAGAAACGCATAGGCCACTAGTGGATCTG |
swd1 5’ genotyping primer |
CTTGCGCACCAGCGTCTC |
swd1 3’ genotyping primer |
CAGCATCTCTTGATGACTGG |
swd3 5’ disruption primer |
CGTAGGCTCATTCATTTGCAAGTGGCCATTATATGCTAACCAGCTGAAGCTTCGTACGC |
swd3 3’ disruption primer |
ACCTTAAGGGAATGTCTGTCGTGTACATAACGCAGATTCTGCATAGGCCACTAGTGGATCTG |
swd3 5’ genotyping primer |
GATACTGTTCCATTTGTCTTTG |
swd3 3’ genotyping primer |
CTTAGAGGAGGTGCATAATTG |
set1 5’ TOPO cloning primer |
ACCATGTCAAATTACTATAGAAGAGC |
set1 3’ TOPO cloning primer |
TCAGTTCAAGAAACCTTTACAAT |
bre2 5’ TOPO cloning primer |
ACCATGAAGTTGGGTATTATACCTTA |
bre2 3’ TOPO cloning primer |
TTATTGCTGTAGGGCAATTTG |
swd1 5’ TOPO cloning primer |
ACCATGAACATCCTTTTACAGGATC |
swd1 3’ TOPO cloning primer |
CTATGATGACTGCATCTTAATTA |
swd3 5’ TOPO cloning primer |
ACCATGTTCCAGTTTGTTACTCCT |
swd3 3’ TOPO cloning primer |
TTAAACCCATCTCCATAAACAG |
MLL1 5’ TOPO cloning primer |
ACCATGGGTACCCCAGATATTGGCTCA |
MLL1 3’ TOPO cloning primer |
TTAGTTTAGGAACTTCCGGCATT |
ASH2L 5’ TOPO cloning primer |
ACCATGGCGGCGGCAGG |
ASH2L 3’ TOPO cloning primer |
TCAGGGTTCCCATGGGGG |
RBBP5 5’ TOPO cloning primer |
ACCATGAACCTCGAGTTGCTGGA |
RBBP5 3’ TOPO cloning primer |
TCATAACAGTTCTGAGATTGCTCC |
WDR5 5’ TOPO cloning primer |
ACCATGGCGACGGAGGAGAAG |
WDR5 3’ TOPO cloning primer |
TTAGCAGTCACTCTTCCACAGTTT |
DPY30 5’ TOPO cloning primer |
ACCATGGAGCCAGAGCAGATGC |
DPY30 3’ TOPO cloning primer |
TCAGTTTCGATCTTCAAACTG |
Constructing the human MLL1 multi-protein complex
All plasmids harboring loxp antibiotic marker cassettes (pUG6, pAG25 and pAG32) and yeast expression plasmids (pAG416 GAL-EGFP-ccdB, pAG413 GAL-EGFP-ccdB, pAG414 GAL-EGFP-ccdB and pAG415 GAL-EGFP-ccdB) used in this study are listed in Table S2. Plasmids harboring loxp antibiotic marker cassettes used for gene deletions were purchased from Euroscarf. Yeast expression plasmids allowing for expression via a galactose inducible promoter were purchased from Addgene (Alberti, Gitler & Lindquist, 2007). All yeast (Set1, Bre2, Swd1, Swd2 and Swd3) and human (MLL1, Ash2L, RbBP5, WDR5 and DPY30) genes were first cloned into pCR8-TOPO (Gateway BP reaction, Invitrogen) according to manufacturer's instructions. The genes were then ligated into yeast expression plasmids using LR clonase (Invitrogen) according to published method [33]. All constructed plasmids were sequenced by Eurofins Operon. All yeast genes and human Ash2L, RbBP5, WDR5 and DPY30 were cloned as full-length constructs into the expression plasmids. A C-terminal fragment of the human MLL1 gene product (amino acids 3370-3969 harboring the WIN and SET domains) was cloned into the yeast expression plasmid.
Table S2. Plasmids used in this study
Plasmid name |
Source |
pUG6 |
(Guldener, et al. 1996) |
pAG25 |
(Goldstein, McCusker, 1999) |
pAG32 |
(Goldstein, McCusker, 1999) |
pRS413-set1-EGFP, HIS |
This study |
pRS415-bre2-EGFP, leu |
This study |
pRS416-swd1-EGFP, URA3 |
This study |
pRS416-swd3-EGFP, URA3 |
This study |
pRS413-MLL1-EGFP, his |
This study |
pRS415-ASH2L-EGFP, leu |
This study |
pRS416-RBBP5-EGFP, URA3 |
This study |
pRS416-WDR5-EGFP, URA3 |
This study |
pRS414-DPY30-EGFP, TRP |
This study |
Yeast Total Protein Extraction for Western Blots
Overnight 5 mL yeast cultures of indicated strains were grown to an ocular density (OD) of 1.5 at 600 nm and subsequently harvested via centrifugation. Pellets were then frozen on dry ice and lysed with 10 M NaOH and 7.4% Betamercaptoethanol. 50% Trichloroacetic Acid was added as previously described [34], cells were washed with Tris and resuspended in 5X SDS buffer. Samples were then loaded on 4-12% NuPAGE Bis-Tris gels (Invitrogen), transferred to PVDF membranes, and then subjected to Western blot analysis using antibodies against histone H3 (Active Motif 61475) acting as a control, H3K4me1, H3K4me2 (Active Motif 39141), and H3K4me3 (Active Motif 39159).
Yeast Growth Assays
For serial dilution yeast growth assays, each yeast strain was plated in a tenfold dilution series to ensure consistent growth pattern and development (i.e. to prove that the genetic altercations have no impact upon the overall yeast growth). In this experiment, the growth assay was as expected, indicating no metabolic or growth-related defects in any yeast strains (Figure 1). Any changes in yeast growth are not attributed to the procedural method involved in specializing the strains and knocking out genes.
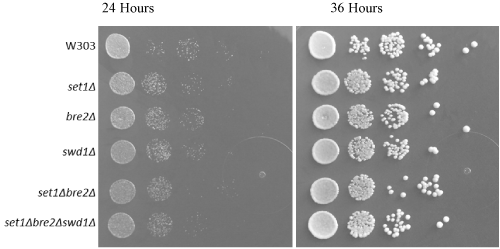
Figure 1. Yeast growth is unchanged with COMPASS gene deletions. Tenfold serial dilution plating showing yeast growth at 24 and 36 hours at 30o C. Each modified strain experienced homogenous growth and therefore growth changes cannot be attributed to the effects of gene deletions
Single Gene Deletions and Replacements
Upon deletion of any core COMPASS member, yeast experienced a global loss of H3K4 methylation. This loss of H3K4 methylation was rescued by expression of the human Bre2 homolog, Ash2L, in the yeast bre2∆ strain (Figure 2). This rescue was the sole replacement capable of restoring partial methylation. The human homologs for Set1 and Swd1 (MLL1 and RbBP5, respectively) were also tested for rescue of H3K4 methylation, but only Ash2L was capable of functioning in place of its COMPASS counterpart, Bre2. MLL1 and RbBP5, the human homologs for Set1 and Swd1, did not rescue di- or trimethylation of H3K4. The Ash2L rescue displayed wildtype-level H3K4 dimethylation and partial trimethylation (Figure 2). Therefore, we have observed that Ash2L can interact with Set1 and Swd1 in the place of Bre2 in the COMPASS complex to form a functional H3K4 di- and tri- methyltransferase.
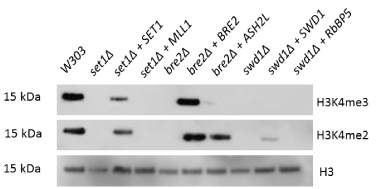
Figure 2. Ash2L can functionally substitute for yeast Bre2, while other single COMPASS gene replacements do not restore H3K4 di- or trimethylation. Yeast strains with individual COMPASS genes deleted were transformed with plasmids encoding either the deleted yeast gene or its human homolog. Total yeast protein extracts from these strains were subjected to Western blotting to detect levels of total histone H3, H3K4 dimethylation and H3K4 trimethylation
Double and Triple Deletions and Replacements
To understand the MLL1 accessory proteins’ abilities to interact with each other and yeast COMPASS members, we created hybrid yeast-human protein complexes by introducing genes encoding human and yeast genes on plasmids into double and triple COMPASS member deletion strains. These chimeric protein complexes serve to reveal the unstudied interactions between accessory proteins of both complexes and hint at the intricate regulatory mechanism surrounding MLL1’s methyltransferase activity. Selected chimeric methyltransferase complexes conferred methyltransferase activity at varying degrees, while others could not form a functional methyltransferase complex.
Assessing the permutations and combinations of the accessory proteins in all yeast strains constructed (Figures 3 and 4) will reveal functional intricacies about the MLL1 protein, its accessory proteins, and maintenance of proper function.
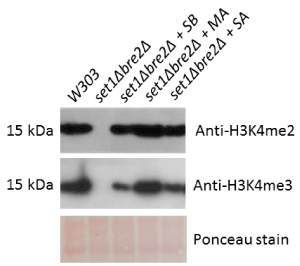
Figure 3. Hybrid yeast-human protein complexes restore H3K4me2 and H3K4me3 to near-wildtype levels in a set1∆bre2∆ background. The set1Δbre2Δ double deletion strain was transformed with multiple plasmids encoding yeast and human gene homologs (SB = Set1 and Bre2, MA = MLL1 and Ash2L, and SA = Set1 and Ash2L). Total yeast protein extracts from these strains were subjected to Western blotting to detect levels of H3K4 dimethylation and H3K4 trimethylation. Ponceau staining was performed as a loading control
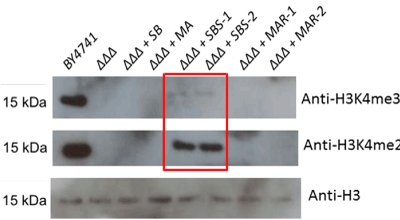
Figure 4. Loss of H3K4 methylation associated with triple COMPASS member deletions cannot be rescued to wildtype levels with either human or yeast methyltransferase members. The set1Δbre2Δswd1Δ triple deletion strain (abbreviated “ΔΔΔ” in the figure) was transformed with plasmids encoding the three deleted yeast genes or their human homologs (SB = Set1 and Bre2, MA = MLL1 and Ash2L, SBS = Set1, Bre2 and Swd1, MAR = MLL1, Ash2L and RbBP5). Total yeast protein extracts from these strains were subjected to Western blotting to detect total histone H3, H3K4 dimethylation and trimethylation
In a set1∆bre2∆ background, rescue of Set1 and Bre2, MLL1 and Ash2L, and Set1 and Ash2L restored dimethylation to wildtype levels and trimethylation to partial levels (Figure 3). These experiments suggest that Set1 and Bre2 are largely interchangeable with their human homologs, MLL1 and Ash2L. Nevertheless, these experiments again demonstrate the essentiality of Set1 and Bre2 to the overall methylation mechanism. Experiments in the double deletion set1Δbre2Δ strain have supported the possibility of certain hybrid interactions, as we have observed a complete rescue of H3K4 dimethylation and a significantly weaker rescue of H3K4 trimethylation in set1Δbre2Δ+Set1+Ash2L. Yeast two-hybrid studies in our lab have confirmed that Ash2L binds to Set1 but their exact interaction remains unknown (data not shown).
Triple rescue of the triple deletion strains resulted in rescue of COMPASS functionality only in the yeast replacement, and not with any combination of human genes (Figure 4).
Chromosomal translocations of the human MLL1 gene have been implicated in a variety of leukemias [2]. Exome sequencing has revealed a much broader suite of MLL1 rearrangements across the gamut of cancers, suggesting that the identification of a drug target within the MLL1 complex could have therapeutic ramifications far beyond treatment of leukemias alone [35]. When translocations occur at the MLL1 locus, the normal function of MLL1 is altered, leading to misregulation of gene expression, including HOX genes [1]. Infant acute lymphoid leukemia patients with MLL1 gene rearrangements show an especially low (15-50%) survival rate compared with those without MLL1 gene rearrangements (60-80%) [36,37].
In order to construct the human MLL1 complex in yeast, we deleted genes encoding Set1, Bre2, and Swd1 from the yeast genome, effectively creating single, double, and triple COMPASS deletion strains. Thereafter, we rescued combinations of these deletions’ human homologs, MLL1, Ash2L, and RbBP5, selected based on the in vitro experiments of Li, et al. [17] and Cao, et al. [18]. We show that human Ash2L may assume the role of Bre2 in yeast COMPASS, thereby suggesting a high degree of capacity for interaction between human and yeast H3K4 methyltransferase complexes. This interactive capacity suggests structural and functional similarities between the complexes and validates the proof of concept presented in this study for use of S. cerevisiae as a model for MLL1 complex function and future drug design work. Recently, two studies have reported structural characterization of yeast COMPASS via cryo-electron microscopy and x-ray crystallography [38,39]. Both structural studied reveal direct interaction of Set1 with Bre2 supporting its integral role in complex activity.
The enzymatic activity of MLL1 is dependent on association with at least three accessory proteins (Ash2L, RbBP5 and WDR5) forming the multi-protein complex [21]. The involvement of these accessory proteins with methyltransferase activity suggests that these proteins play a crucial role in regulating the enzymatic activity of MLL1. Uncovering the specific roles of these accessory proteins may illuminate new drug targets for treatment of leukemia and other cancers involving MLL1 translocations that render the MLL1 enzyme itself a problematic. The chromosomal translocations involving the human MLL1 gene result in fusion proteins where a portion of MLL1 is fused to numerous other proteins [40]. The fact that MLL1 is often altered in a dramatic and inconsistent way among patients with leukemia renders MLL1 a poor target of directed leukemia therapies. However, since MLL1 functions within the context of a multi-protein complex, uncovering the roles fulfilled by accessory proteins in regulating MLL1 enzymatic activity may implicate a specific complex member as new drug target for leukemia. Site-directed mutagenesis experiments to introduce specific known mutations in MLL1 complex members may alter MLL1 activity in the yeast strain, thereby implicating the member as a potential drug target. The results of future site-directed mutagenesis experiments will help elucidate the role of accessory proteins in regulation of the enzymatic activity of the complex and potentially identify drug targets within the multi-protein MLL1 complex.
This publication was supported by grants from the National Center for Research Resources (5 P20 RR016461) and the National Institute of General Medical Sciences (8 P20 GM103499) from the National Institutes of Health.
The authors declare no conflicts of interest.
- Argiropoulos B, Humphries RK (2007) Hox genes in hematopoiesis and leukemogenesis. Oncogene 26: 6766-6776. [Crossref]
- Harper DP, Aplan PD (2008) Chromosomal rearrangements leading to MLL gene fusions: clinical and biological aspects. Cancer research 68: 10024-10027. [Crossref]
- Schneider J, Wood A, Lee JS, Schuster R, Dueker J, et al. (2005) Molecular regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Mol Cell 19: 849-856. [Crossref]
- Slany RK (2009) The molecular biology of mixed lineage leukemia. Haematologica 94: 984-993. [Crossref]
- Muntean AG, Hess JL (2012) The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol 7: 283-301. [Crossref]
- Wiederschain,D, Kawai H, Shilatifard A, Yuan ZM (2005) Multiple mixed lineage leukemia (MLL) fusion proteins suppress p53-mediated response to DNA damage. The Journal of biological chemistry 280: 24315-24321. [Crossref]
- Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M (2012) Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov 11: 384-400. [Crossref]
- Wu L, Lee SY, Zhou B, Nguyen UT, Muir TW, et al. (2013) ASH2L regulates ubiquitylation signaling to MLL: trans-regulation of H3 K4 methylation in higher eukaryotes. Mol Cell 49: 1108-1120. [Crossref]
- Steward MM, Lee JS, O'Donovan A, Wyatt M, Bernstein BE, et al. (2006) Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol. 13: 852-854. [Crossref]
- Butler JS, Zurita-Lopez C, Clarke SG, Bedford MT, Dent SY (2011) Protein-arginine methyltransferase 1 (PRMT1) methylates Ash2L, a shared component of mammalian histone H3K4 methyltransferase complexes. J Biol Chem 286: 12234-12244. [Crossref]
- Patel A, Vought VE, Swatkoski S, Viggiano S, Howard B, et al. (2014) Automethylation activities within the mixed lineage leukemia-1 (MLL1) core complex reveal evidence supporting a "two-active site" model for multiple histone H3 lysine 4 methylation. J Biol Chem 289: 868-884. [Crossref]
- Zhang P, Chaturvedi CP, Tremblay V, Cramet M, Brunzelle JS, et al. (2015) A phosphorylation switch on RbBP5 regulates histone H3 Lys4 methylation. Genes Dev 29: 123-128. [Crossref]
- Karatas H, Li Y, Liu L, Ji J, Lee S, et al. (2017) Discovery of a Highly Potent, Cell-Permeable Macrocyclic Peptidomimetic (MM-589) Targeting the WD Repeat Domain 5 Protein (WDR5)-Mixed Lineage Leukemia (MLL) Protein-Protein Interaction. J Med Chem 60: 4818-4839. [Crossref]
- Cao F, Townsend EC, Karatas H, Xu J, Li L, et al. (2014) Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell 53: 247-261. [Crossref]
- Grebien F, Vedadi M, Getlik M, Giambruno R, Grover A, et al. (2015) Pharmacological targeting of the Wdr5-MLL interaction in C/EBPalpha N-terminal leukemia. Nat Chem Biol 11: 571-578. [Crossref]
- Alicea-Velazquez NL, Shinsky SA, Loh DM, Lee JH, et al. (2016) Targeted Disruption of the Interaction between WD-40 Repeat Protein 5 (WDR5) and Mixed Lineage Leukemia (MLL)/SET1 Family Proteins Specifically Inhibits MLL1 and SETd1A Methyltransferase Complexes. J Biol Chem 291: 22357-22372. [Crossref]
- Li Y, Han J, Zhang Y, Cao F, Liu Z, et al. (2016) Structural basis for activity regulation of MLL family methyltransferases. Nature 530: 447-452. [Croosref]
- Cao F, Chen Y, Cierpicki T, Liu Y, Basrur V, et al. (2010) An Ash2L/RbBP5 heterodimer stimulates the MLL1 methyltransferase activity through coordinated substrate interactions with the MLL1 SET domain. PloS one 5: e14102. [Crossref]
- Shinsky SA, Monteith KE, Viggiano S, Cosgrove MS (2015) Biochemical reconstitution and phylogenetic comparison of human SET1 family core complexes involved in histone methylation. J Biol Chem 290: 6361-6375. [Crossref]
- Ruthenburg AJ, Allis CD, Wysocka J (2007) Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell 25: 15-30. [Crossref]
- Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, et al. (2006) Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol 13: 713-719. [Crossref]
- Tuukkanen A, Huang B, Henschel A, Stewart F, Schroeder M (2010) Structural modeling of histone methyltransferase complex Set1C from Saccharomyces cerevisiae using constraint-based docking. Proteomics 10: 4186-4195. [Crossref]
- Takahashi YH, Westfield GH, Oleskie AN, Trievel RC, Shilatifard A, et al. (2011) Structural analysis of the core COMPASS family of histone H3K4 methylases from yeast to human. Proc Natl Acad Sci U S A 108: 20526-20531. [Crossref]
- Zhang H, Li M, Gao Y, Jia C, Pan X, et al. (2015) Structural implications of Dpy30 oligomerization for MLL/SET1 COMPASS H3K4 trimethylation. Protein & cell 6: 147-151. [Crossref]
- Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, et al. (2001) COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci U S A 98: 12902-12907. [Crossref]
- Dehe PM, Dichtl B, Schaft D, Roguev A, Pamblanco M, et al. (2006) Protein interactions within the Set1 complex and their roles in the regulation of histone 3 lysine 4 methylation. J Biol Chem 281: 35404-35412. [Crossref]
- Takahashi YH, Westfield GH, Oleskie AN, Trievel RC, Shilatifard A, et al. (2011b) Structural analysis of the core COMPASS family of histone H3K4 methylases from yeast to human. Proc Natl Acad Sci U S A 108: 20526-20531. [Crossref]
- Zhang K, Lin W, Latham JA, Riefler GM, Schumacher JM, et al. (2005) The Set1 methyltransferase opposes Ipl1 aurora kinase functions in chromosome segregation. Cell 122: 723-734. [Crossref]
- Amberg DC, Burke DJ, Strathern JN (2005) Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual, 2005 Edition (Cold Spring).
- Goldstein AL, McCusker JH (1999) Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15: 1541-1553. [Crossref]
- Güldener U, Heck S, Fielder T, Beinhauer J, Hegemann JH (1996) A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res 24: 2519-2524. [Crossref]
- Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH (2002) A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30: e23. [Crossref]
- Alberti S, Gitler AD, Lindquist S (2007) A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast (Chichester, England) 24: 913-919. [Crossref]
- Ooi CE, Rabinovich E, Dancis A, Bonifacino JS, Klausner RD (1996) Copper-dependent degradation of the Saccharomyces cerevisiae plasma membrane copper transporter Ctr1p in the apparent absence of endocytosis. EMBO J 15: 3515-3523. [Crossref]
- Rao RC, Dou Y (2015) Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer 15: 334-346. [Crossref]
- Chen CW, Armstrong SA (2015) Targeting DOT1L and HOX gene expression in MLL-rearranged leukemia and beyond. Exp Hematol 43: 673-684. [Crossref]
- Hilden JM, Dinndorf PA, Meerbaum SO, Sather H, Villaluna D, et al. (2006) Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children's Oncology Group. Blood 108: 441-451. [Crossref]
- Hsu PL, Li H, Lau HT, Leonen C, Dhall A, et al. (2018) Crystal Structure of the COMPASS H3K4 Methyltransferase Catalytic Module. Cell 174: 1106-1116.e9. [Crossref]
- Wang Y, Ding Z, Liu X, Bao Y, Huang M, et al. (2018) Architecture and subunit arrangement of the complete Saccharomyces cerevisiae COMPASS complex. Sci Rep 8:17405-018-35609-8. [Crossref]
- Ballabio E, Milne TA (2012) Molecular and Epigenetic Mechanisms of MLL in Human Leukemogenesis. Cancers (Basel) 4: 904-944. [Crossref]