In the past, interactions between drugs and vitamin D have received only little or no attention in the health care practices. However, since more and more drugs are used for the treatment of patients, this topic is increasingly relevant. Several drugs can interfere with the vitamin D and bone metabolism. Drugs that activate the pregnane X receptor can disrupt vitamin D metabolism and vitamin D function. Beside this, the medication oriented supplementation of vitamin D can ameliorate the pharmacologic action of some drugs, such as bisphosphonates, cytostatics and statins.
Vitamin D plays a crucial role in human health, while the prevalence of vitamin D deficiency worldwide is high. According to recent studies, a vitamin D deficiency is likely to be an important etiological factor in the pathogenesis of many chronic diseases. These include autoimmune diseases (e.g., multiple sclerosis, type 1 diabetes) inflammatory bowel disease (e.g., Crohn disease), infections (such as infections of the upper respiratory tract), immune deficiency, cardiovascular diseases (e.g., hypertension, heart failure, sudden cardiac death), cancer (e.g., colon cancer, breast cancer, non-Hodgkin’s lymphoma) and neurocognitive disorders (e.g., Alzheimer disease). Vitamin D in its hormonally active form, 1α,25-dihydroxyvitamin D [1α,25(OH)2D; calcitriol] is not only a regulator of calcium and phosphate homeostasis, but has numerous extra-skeletal effects. 1α,25(OH)2D manifests its diverse biological effects (endocrine, autocrine, paracrine) by binding to the vitamin D receptor (VDR) found in most body cells. Vitamin D receptors have been found in over 35 target tissues that are not involved in bone metabolism. These include endothelial cells, islet cells of the pancreas, hematopoietic cells, cardiac and skeletal muscle cells, monocytes, neurons, placental cells and T-lymphocytes. It is estimated that VDR activation may regulate directly and/or indirectly a very large number of genes (0.5–5% of the total human genome i.e., up to 6000 genes). The fact that the vitamin D receptor is expressed by many tissues results in the pronounced pleiotropic effect of vitamin D hormone. 25(OH)D is the vitamin D metabolite that is measured to assess a patient’s vitamin D status. Vitamin D deficiency is diagnosed when 25(OH)D <20 ng/mL, vitamin D insufficiency is defined as 25(OH)D of 21–29 ng/mL, and 25(OH)D > 30 ng/mL is considered sufficient, with 40–60 ng/mL being the preferred range. Vitamin D intoxication usually does not occur until 25(OH)D >150 ng/mL. Vitamin D intoxication is only to be expected at levels of 25(OH)D > 150 ng/mL [1-3].
Interactions between drugs and vitamin D have received only little or no attention in the medical and pharmaceutical world in the past. Since more and more drugs are used for the treatment of patients, this topic is increasingly relevant. As such interactions impact the health of the patient and the action and side effects of the drug, physicians and pharmacists should pay more attention to this fact in the future. As vitamin D deficiency leads to bone damage, it is particularly important to ensure an adequate vitamin D supply in cases of pre-existing osteoporosis or during long-term intake of drugs that promote the development of bone damage. Even after bone damage has already occurred, therapeutic use of vitamin D is often not considered [4,5].
A number of drugs are known to interfere with the vitamin D metabolism through activation of the pregnane X receptor and thereby causing vitamin D deficiency. Through prevention or treatment of vitamin D deficiency, the risk of drug-induced bone damage, such as that caused by antiepileptic agents, glucocorticoids, anti-estrogens or antiretroviral drugs, can be reduced. For adequate bisphosphonate response in osteoporosis therapy a sufficient vitamin D status must be ensured. Initial studies indicate that vitamin D also has an effect on the lipid-lowering activity of statins (HMG-CoA reductase inhibitors) and the antibacterial effect of antituberculotic agents. The following article discusses the mechanisms of an interaction between vitamin D and the relevant drug groups. In many cases, monitoring of serum 25-hydroxy-vitamin D [25(OH)D] levels and compensation of vitamin D deficiency can contribute to reducing the risk of adverse drug reactions and/or improving the efficacy of various drugs.
Pregnane X receptor mediated interactions
Vitamin D from the skin and diet is metabolized in the liver to 25-hydroxy-vitamin D [25(OH)D]. 25(OH)D is the major circulating form of vitamin D and is used to determine a patient’s vitamin D status. 25(OH)D is metabolized in the kidneys by the enzyme 25-hydroxyvitamin D-1α-hydroxylase (CYP27B1) to its active form, 1α,25-dihydroxy-vitamin D [1,25(OH)2D]. Both 25(OH)D and 1,25(OH)2D are oxidized by hydroxylases (CYP24A1, CYP3A4) at position 24 of the side chain. The resultant metabolites are physiologically inactive and are excreted as acids following further metabolic stages. Expression of the 24-hydroxylases is partially dependent on the calcium and parathyroid hormone levels in the blood and partially regulated by 1,25(OH)2D itself. In this way, the concentration of circulating 1,25(OH)2D and thus both calcium and phosphate homeostasis in the blood is strictly regulated.
Various drugs can interfere in this balance through activation of the pregnane X receptor (PXR). In 1998 the pregnane X receptor (PXR) of mouse was first identified as a member of the nuclear receptor (NR) superfamily on the basis of its sequence homology with other NRs. Human PXR (hPXR) was found subsequently and named steroid and xenobiotic receptor (SXR) or pregnane activated receptor. The Pregnane X receptor (PXR) plays an important role in detoxifying xenobiotics and drugs. It is an intracellular receptor, which is expressed in the cells of the gastrointestinal tract, the kidneys and the liver and shows 60% homology with the vitamin D receptor in the DNA-binding domains. The pregnane X receptor can thereby bind to vitaminD-responsive elements (VDRE) at the DNA and, as a transcription factor, affect the expression of genes whose expression is normally regulated by vitamin D. PXR-ligands are structurally diverse and include a wide variety of pharmaceutical agents, such as antiepileptics (e.g. phenytoine), anti-inflammatory agents, antiretroviral drugs, cytostatics (e.g. paclitaxel) (Table 1).
Table 1. Drugs that activate the pregnane-X-receptor (PXR) (selection)
|
PXR-Ligands Examples
|
PXR-Ligands Examples
|
|
Antiepileptics
|
Phenytoin, carbamazepine
|
|
Antineoplastic drugs
|
Cyclophophamide, epirubicin, taxol,
tamoxifen
|
|
Antibiotics
|
Clotrimazole, rifampicin
|
|
Anti-inflammatory agents
|
Dexamethasone
|
|
Antihypertensives
|
Nifedipine, spironolactone
|
|
Antiretroviral drugs
|
Efavirenz, ritonavir, saquinavir
|
|
Endocrine drugs
|
Cyproterone acetate
|
Commonly used herbal medicines can also activate PXR, such as St John’s wort and kava kava. Through activation of the pregnane X receptor, expression of the 24-hydroxylases is upregulated, leading to increased degradation of 25(OH)D and 1,25(OH)2D (Figures 1). It is still unclear whether other effects of vitamin D are “imitated” by activation of the pregnane X receptor. In addition to the 24-hydroxylases, the ligands of the pregnane X receptor induce other cytochrome P450 enzymes, which are involved in the biotransformation of numerous active substances (e.g., CYP2C9 and CYP3A4).
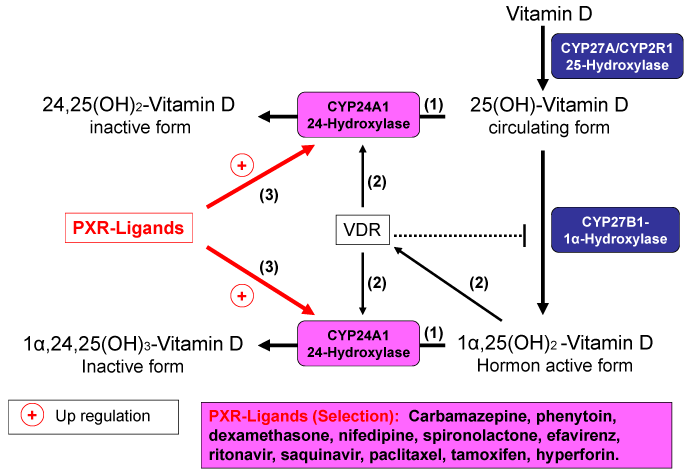
Figure 1. Induction of 24-Hydroxylase by PXR-Ligands and distruction of vitamin D
Antiepileptic drugs
It was documented more than 40 years ago that institutionalized children who were on multiple anti-seizure medications developed rickets that was resistant to normal vitamin D therapy. As a result of their disease and the associated tendency to fall, patients with epilepsy are at higher risk of bone fractures. In addition, many antiepileptic drugs (AEDs) promote the pathogenesis of AED-induced bone disease, which is detected in up to 50% of patients undergoing long-term antiepileptic treatment. The risk of bone fractures is two to six times higher in patients with epilepsy than in the average population and comparable to that seen in patients undergoing long-term glucocorticoid therapy
AED-induced disturbances of bone integrity are mainly influenced by the type, dosage and duration of the antiepileptic therapy. A dose-dependent increase in the risk of fractures was particularly observed during therapy with carbamazepine, oxcarbazepine, clonazepam, Phenobarbital, phenytoin, primidone, and valproic acid. The risk of AED-induced bone disease was greater with inducers of cytochrome P450 (CYP), i.e. carbamazepine, phenobarbital, phenytoin and primidone, than with other antiepileptic agents.
AED-induced disturbances of bone metabolism are usually accompanied by a fall in the 25(OH)D level, hypocalcemia, secondary hyperparathyroidism, and increased bone turnover with a decrease in bone density. In the pathogenesis of AED-induced bone disease, a central role is played by the pharmacokinetic interaction between the AEDs and vitamin D: the enzyme inducers carbamazepine, phenobarbital, phenytoin, and primidone can activate the pregnane X receptor, which then upregulates expression of the 24-hydroxylases, which can cause vitamin D deficiency.
AED-induced bone disease can also occur even with more modern AEDs, such as gabapentin, lamotrigine and levetiracetam, which have little or no effect on the activity of the cytochrome enzyme, as other mechanisms are probably also involved in the development of this bone damage. Valproic acid-induced osteopathy, for example, cannot be explained by an induction of 24-hydroxylases, as valproic acid inhibits cytochrome P450 enzymes and is not a ligand of the pregnane X receptor.
Prophylaxis with vitamin D is recommended for all subjects using AEDs. Due to increased catabolism of vitamin D, higher than normally recommended doses (up to 7000 IU per day) of vitamin D are required for optimal effect, particularly for those with low vitamin D levels, high risk of bone disease and/or with documented low bone mineral density (BMD). In general, in patients undergoing antiepileptic treatment, vitamin D status should be monitored once to twice annually, based on the serum 25(OH)D level (target: 40–60 ng/mL [100-150 nmol/L]). Any deficiency should be treated as required with targeted supplementation in order to prevent osteopathy. For those with documented vitamin D deficiency, treatment with 40,000 IU vitamin D/per day for 2 weeks is recommended, followed thereafter by 5000 IU of vitamin D every day.
Antiretroviral drugs
In persons infected with the human immunodeficiency virus (HIV), the osteoporosis risk is more than three times higher than in persons not infected with HIV. The increased risk in HIV-infected persons is partially due to the fact that the HI virus impairs bone integrity. It has been shown that glycoproteins of HIV-1 (p55-gag, gp120) impair bone calcium utilization and reduce osteoblast activity. In infected macrophages, HIV-1 induces the production of macrophage colony-stimulating factor (M-CSF), which, together with the RANK ligand, increases osteoclastogenesis. Upregulation of proinflammatory cytokines (e.g. tumor necrosis factor alpha [TNF-α]) can additionally induce osteoblast apoptosis and thus further increase the risk of viral damage to the bone cells.
The risk of osteopathy is additionally increased by antiretroviral therapy. Disturbances of vitamin D metabolism, particularly an increased vitamin degradation due to induction of CYP3A4, appear to play a major role. Vitamin D deficiency is frequently observed in HIV-infected patients: in a study with 1,077 HIV-infected patients, 91% of subjects had a suboptimal 25(OH)D level and one third actually had severe vitamin D deficiency (25(OH)D <10 ng/mL). In this study, the risk of severe vitamin D deficiency was significantly increased by intake of the non-nucleoside reverse transcriptase inhibitor efavirenz. In individuals infected with HIV, vitamin D deficiency has negative effects not only on the bones, but also on viral load and disease progression.
Against this background, vitamin D administration in individuals infected with HIV appears appropriate, in order to reduce the risk of drug-induced osteopathy. Vitamin D may also reduce the mitochondrial toxicity of antiretroviral virostatic drugs, whose effects include muscle pain and lipid metabolism disorders. For those with documented vitamin D deficiency, treatment with 40,000 IU vitamin D/per day for 2 weeks is recommended, followed thereafter by 50 IU vitamin D daily per kg bodyweight.
Glucocorticoids
Various factors contribute to the development of glucocorticoid-induced osteoporosis: glucocorticoids increase osteoclast activity through raised expression of RANK (receptor activator of [nuclear factor kappa B] NFkB) ligand and a reduced production of osteoprotegerin and they reduce the development and differentiation of osteoblasts. Furthermore, glucocorticoids reduce the production of sex hormones, thereby reducing their positive effect on the bones. Glucocorticoids also reduce intestinal calcium absorption and concurrently increase renal calcium excretion; this can lead to a fall in serum calcium levels. Glucocorticoids thus influence the function of osteoblasts and osteoclasts via various direct and indirect effects and some of these effects counteract those of vitamin D. Some glucocorticoids (e.g. dexamethasone) also cause increased degradation of 25(OH)D and 1,25(OH)2D due to activation of the pregnane X receptor.
In patients with multiple sclerosis high-dose and short-term intravenous glucocorticoid regimens can cause a decrease in bone formation. Multiple sclerosis (MS) is generally associated with reduced bone mass and higher frequency of osteoporosis. The results of a small study with 41 women on glucocorticoid therapy, who were recently diagnosed with systemic lupus erythematodes, multiple sclerosis, rheumatoid arthritis or asthma bronchiale indicate, that 1-alpha-hydroxycholecalciferol (0,5-1,0 µg/d) treatment appears to be effective in preventing glucocorticoid-induced bone loss by reducing secondary hyperparathyroidism and stimulating bone formation.
During long- and short-term glucocorticoid therapy, the vitamin D status should always be monitored, especially in patients with multiple sclerosis and bronchial asthma, by means of laboratory tests and any deficiency corrected by means of targeted supplementation, in order to reduce the risk of glucocorticoid-induced disturbances of bone metabolism.
Beyond that, clinical evidence suggests an important role of vitamin D deficiency as a modifiable risk factor in MS. Low circulating levels of 25(OH)D have been found in MS patients, especially during relapses, suggesting that vitamin D could be involved in the regulation of the clinical disease activity. 1,25(OH)2D, the most important form of vitamin D metabolically, possesses pronounced anti-inflammatory and immunomodulatory properties. In MS patients Vitamin D as an add on therapy to interferon β-1b has been shown to reduce MRI disease activity.
Patients undergoing glucocorticoid treatment of bronchial asthma could derive a further benefit from vitamin D supplementation. Patients with low 25(OH)D levels suffered considerably more often from respiratory infections than patients with normal 25(OH)D levels. In a study with children suffering from bronchial asthma, the association between 25(OH)D levels, lung function and the antiasthmatic medicine was investigated. The lower the 25(OH)D levels, the poorer the lung function values of the children and the higher the glucocorticoid doses with which the children were treated. Corticosteroid use and worsening airflow limitation were associated with lower 25(OH)D serum levels in asthmatic patients. A possible explanation for this is that 1,25(OH)2D modulates the expression of cytokines with marked anti-inflammatory and anti-allergic properties (e.g. interleukin 10). In-vitro studies also showed that vitamin D can enhance the immunosuppressive function of dexamethasone.
Cholesterol lowering drugs, statins
Several studies have shown a potential role of vitamin D for prevention and treatment of statin-associated muscle symptoms (SAMS). The prevalence of myalgia in statin-treated patients vitamin D insufficiency is significantly higher compared with those that not present any signs of SAMS. In a trial with 82 vitamin-D-deficient, myalgic patients, under statin therapy, 38 were given vitamin D (50,000 units/weekfor 12 weeks), with a resultant increase in serum 25(OH)D from 20.4 +/27.3 to 48.2 +/217.9 ng/mL (P<0.0001) and resolution of myalgia in 35 (92%). The results of a retrospective cohort study and a meta-analysis of 7 studies with 2420 patients provide evidence that low vitamin D levels are associated with myalgia in statin treated patients. A recent study with 74 men and 72 women (age 59 ± 14 years) reported that statin intolerance because of myalgia, myositis, myopathy, or myonecrosis associated with low serum vitamin D can be safely resolved by vitamin D supplementation (50,000-100,000 units /week) in most cases (88-95%). Vitamin D insufficiency can lead to the development of non-specific muscle pain and SAMS. In older adults vitamin D supplementation even seems to improve the adherence to and persistence with long-term statin treatment. But the association between low vitamin D levels and statin-associated muscle symptoms is still conflicting. Some retrospective cohort studies do not support an association between low 25(OH)D levels and statin-induced myalgia. Given the major clinical importance of SAMS, the results of a large double-blind, placebo-controlled crossover study are needed to elucidate the validity of vitamin D supplementation in ameliorating muscle symptoms on statin therapy.
Recommendation for clinical practice: Although previous results of vitamin D treatment of SAMS from randomized trials have produced mixed results, Vitamin D deficiency is common in statin users and evident in European population at prevalence rates that are concerning. Therefore vitamin D status [25(OH)D, ng/mL) should be monitored in all statin-treated patients and compensated by adequate vitamin D supplementation [e.g., 40–60 IU vitamin D per kg body weight per day, 25(OH)D target value: 40–60 ng/mL or 100–150 nmol/L]. This applies, in particular to patients with cardiovascular diseases, diabetes, the elderly (> 60 years) with poor nutritional status, and statin treated patients with muscular disorders (Figure 2) [6].
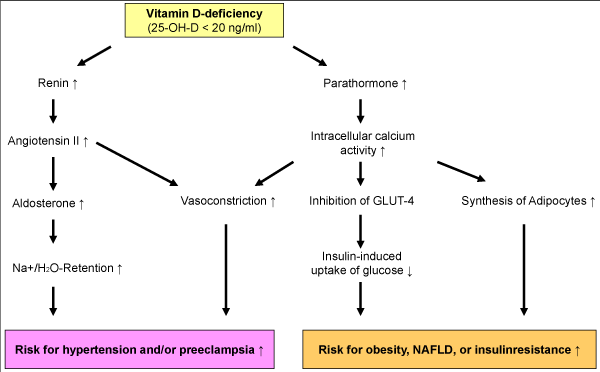
Figure 2. Vitamin D-deficiency and development of hypertension and insulin resistance (possible mechanisms)
The efficacy and side effect rate of several drugs can be improved by vitamin D. With regard to pharmacokinetic interactions, mediated by the pregnane X receptor, it can be assumed that the active substances described in this paper are not the only ones that interact with the PXR-VDR system and can lead to vitamin D deficiency. During long-term medication, therefore, vitamin D status (serum 25(OH)D level) should generally be monitored and any deficiency corrected. Measurement of vitamin D status and subsequently targeted, individual vitamin D supplementation is advisable for preventative and supportive reasons in many diseases and drug therapies [7].
- 1. Gröber U, Reichrath J, Holick MF (2015) Live longer with vitamin D? Nutrients 7: 1871-1880.
- 2. Holick, MF (2007) Vitamin D deficiency. N Engl J Med 357: 266-281.
- 3. Wolf G (2004) The discovery of vitamin D: the contribution of Adolf Windaus. J Nutr 134: 1299-1302.
- 4. Gröber U, Holick MF (2020) Vitamin D – die Heilkraft des Sonnenvitamins. 4. Auflage, Wissenschaftliche Verlagsgesellschaft, Stuttgart.
- 5. Gröber U, Kisters, K (2012) Influence of drugs on vitamin D and calcium metabolism. Dermatoendocrinol 4: 158-166.
- 6. Gröber U, Kisters K, Schmidt J (2018) Important drug-micronutrient interactions: A selection for clinical practice. Crit Rev Food Sci Nutr 23:1-19.
- 7. Gröber U (2020) Common drugs as micronutrient disruptors: A selection for clinical practice. Ann Epidemiol Public Health 3: 1014-1031.