Around 15-20% of human breast cancers are classified as triple-negative (TNBC) because they are devoid of the three common chemotherapy targets. Since current protocols to treat TNBC are largely ineffective, new therapies for TNBC need to be identified. Almost 80% of TNBCs express a functionally defective form of the p53 tumor suppressor protein (mutant p53; mtp53). In mtp53-expressing cancers, most p53 mutations occur in the DNA-binding domain, blocking normal regulation of p53 target genes that are involved in apoptosis, cell-cycle arrest, and angiogenesis and resulting in resistance to chemotherapy and metastasis. Restoring p53 function is therefore a potentially effective strategy for combating TNBC. APR-246 is a small-molecule drug that has been shown to reactivate mtp53 and restore p53 function. We examined whether APR-246 could inhibit TNBC growth in vitro and in vivo. TNBC growth in vitro was measured using cell viability assays, and TNBC growth in vivo was assessed using a tumor xenograft mouse model. Nuclear extracts of APR-246-treated MDA-MB-231 TNBC cells exhibited significantly increased DNA binding compared with untreated cells, indicating that APR-246 converts mtp53 to a wild-type p53 form (wtp53) in these cells. APR-246 significantly reduced viability of MDA-MB-231 and MDA-MB-468 TNBC cells in vitro, but had no effect on normal mammary cells (AG11132A) or breast cancer cells that express wtp53 (MCF-7). In our tumor xenograft mouse model, administration of APR-246 alone or in combination with 2aG4, an antibody that disrupts tumor vasculature, significantly reduced TNBC tumor growth (MDA-MB-231), as well as two markers of angiogenesis (tumor vascular endothelial growth factor expression and blood-vessel density). APR-246 in combination with 2aG4 completely eradicated almost 20% of the TNBC tumors present prior to treatment. Thus, in conclusion, APR-246 alone and in combination with 2aG4 inhibits TNBC tumor growth, and could represent an innovative therapy for TNBC, for which few effective treatment options are currently available.
breast cancer, TNBC tumors, blood-vessel targeting agent, p53, APR-246
ANOVA: analysis of variance; ELISA: enzyme-linked immunosorbent assay; FACS: fluorescence-activated cell sorting; FBS: fetal bovine serum; FITC: fluorescein isothiocyanate; ip: intraperitoneal; iv: intravenous; mtp53: mutant form of p53 tumor suppressor protein; PBS: phosphate-buffered saline; PI: propidium iodide; SEM: standard error of the mean; SRB: sulforhodamine B; TBS-T: Tris-buffered saline with 0.1% Tween 20; TNBC: triple-negative breast cancer; VEGF: vascular endothelial growth factor; wtp53: wild-type p53 tumor suppressor protein
Triple-negative breast cancers (TNBC) constitute approximately 15-20% of all detected human breast cancers. TNBC fail to express estrogen receptor; progesterone receptor; and Her-2-neu [1-4], the three molecules commonly targeted chemotherapeutically in hormone receptor-positive tumors. Although aggressive non-targeted chemotherapeutic approaches are typically used against TNBC, such protocols are generally ineffective, making this cancer virtually untreatable. In addition, TNBC tumors frequently metastasize, leading to poor patient prognosis and death. It is therefore important to develop better treatment strategies to reduce TNBC-related breast cancer mortality, since for patients with this aggressive type of cancer, the prognosis is not favorable following recurrence.
While assessing the genome of TNBC tumors, it was recently discovered that nearly 80% of TNBCs express an inactive mutant form of the p53 tumor suppressor protein (mtp53) [5]. The function of wild-type p53 tumor suppressor protein (wtp53) is to promote cell-cycle arrest and apoptosis and to inhibit vascular endothelial growth factor (VEGF)-dependent angiogenesis, which, if left unchecked, causes rapid tumor growth, metastasis, and mortality [6-12]. The majority of p53 mutations occur in the DNA-binding domain, which leads to aberrant regulation of p53 target genes involved in the aforementioned processes of apoptosis, cell-cycle arrest, and/or angiogenesis [13,14]. Dysregulation of these processes results in neovascularization, unrestricted tumor growth and metastasis, and can lead to drug resistance [15]. Further, wtp53 curbs both the self-renewal properties of stem cells and the epithelial-to-mesenchymal transition, the latter of which plays a vital role in initiating tumor metastasis, which leads ultimately to patient death [16-18]. We contend therefore that restoring functional wtp53 in women who suffer from p53-defective TNBC may be a feasible alternative therapeutic approach to treat this aggressive type of cancer. In addition, restoring wtp53 function may allow lower levels of chemotherapeutic agents to be administered to these patients, given that wtp53 is known to enhance chemotherapeutic effects.
Our recent study identified APR-246 as a useful compound in preventing lung metastasis of TNBC cells in an animal model [19]. APR-246 re-activates mtp53 by covalent modification of the DNA-binding core domain of the mutant protein through alkylation of thiol groups [20,21], thus restoring wild-type conformation and function. APR-246 has been found to restore wtp53 conformation in mtp53-expressing breast cancer cells and to renew its capacity to promote cell-cycle arrest and apoptosis in these cells [22,23]. Given these findings, we sought to determine whether APR-246 inhibits growth of TNBC tumor cells, thereby preventing the development of primary tumors.
Herein, we examined the effects of APR-246 on TNBC cells in vitro. We also assessed the therapeutic effectiveness in vivo of combining APR-246 with the 2aG4 antibody (human equivalent bavituximab; U.S. Food and Drug Administration-approved for clinical trials). 2aG4 targets exposed phosphatidylserine residues on tumor blood vessels, blocks vessel function, and reduces tumor angiogenesis [24-26]. We found that, in vitro, APR-246 reduced TNBC cell viability, induced TNBC cell apoptosis, and inhibited secretion of VEGF from TNBC cells. In our in vivo studies using a tumor xenograft mouse model, APR-246 alone and in combination with 2aG4 inhibited TNBC tumor growth, as well as reduced VEGF expression and blood-vessel density in TNBC tumor tissues. No animal toxicity was observed following treatment with either agent alone or in combination. Further, treatment with APR-246 alone, or a combination of APR-246 and 2aG4, completely eradicated some TNBC tumors in the timeframe tested. These findings suggest that APR-246 alone and in combination with immunotherapy has the capacity to inhibit TNBC and could represent a novel therapy to stop the emergence of drug-resistant tumors in TNBC patients.
Cell lines and culture
All cell-culture studies were approved by the University of Missouri Institutional Environmental Health and Safety Board (Columbia, MO, USA). MDA-MB-231 (Basal B) and MDA-MB-468 (Basal A) TNBC cells, as well as hormone receptor-positive MCF-7 cells, were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were grown in DME/F12 medium supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO, USA) at 37oC in a humidified atmosphere of 5% CO2 and harvested for different experiments with 0.05% trypsin-EDTA (ThermoFisher Scientific, Waltham, MA, USA). Cells were washed with phosphate-buffered saline (PBS) or FBS-free medium before being treated in fresh medium containing 5% FBS. AG11132A normal mammary cells were obtained from the American Type Culture Collection and grown as specified by the vendor.
Reagents
APR-246 (PRIMA-1MET) was obtained from Tocris Bioscience (Bristol, UK). APREA AB (Solinka, Sweden) kindly provided the APR-246 used in in vivo studies. 2aG4, a mouse IgG2a monoclonal antibody that binds directly to phosphatidylserine residues on tumor blood vessels [27] was provided by Dr. Rolf Brekken from the University of Texas Southwestern Medical Center, Dallas, TX. Binding of 2aG4 to anionic phospholipids depends on a 50-kD bovine plasma glycoprotein, β2-glycoprotein 1. We therefore mixed 2aG4 with β2-glycoprotein 1 at a 1:1 ratio to enhance binding of 2aG4 to anionic phospholipids exposed on the endothelial cell surface [28]. C44, the IgG2a mouse anti-colchicine monoclonal antibody (also provided by Dr. Brekken) was used as a negative control for 2aG4.
Activation of p53 DNA binding
Conformation and activation of p53 were analyzed using a TransAM p53 Transcription Factor Assay kit (Active Motif, Carlsbad, CA, USA) as described before [29]. 96-well plates were coated with an oligonucleotide containing a p53 consensus DNA-binding site. An extraction kit (Active Motif) was used to prepare nuclear extracts from MDA-MB-231 cells. 2.5 µg of nuclear extract was added to the 96-well plates and incubated with the aforementioned oligonucleotide. Bound p53 was detected by the addition of anti-p53 antibody together with a secondary antibody conjugated to horseradish peroxidase (both antibodies at dilutions of 1:1000). A Spectra MAX 190 Microplate Reader (Molecular Devices, Sunnyville, CA, USA) was employed to measure developed color at 450 nm. Background OD was established using a 20-fold excess of free oligonucleotide and the background value was subtracted from total binding to generate specific binding values. MCF-7 nuclear extract treated with H2O2 was used as a positive control.
Cell viability assay
Cell viability was determined by the sulforhodamine B assay (SRB, Sigma-Aldrich) [30,31]. The SRB assay quantitates protein content of surviving cells as an index of cell growth and viability, as described in our previous publications [29,30]. Six wells were used for each concentration, and experiments were performed three times.
Apoptosis and cell death assay
Apoptosis and cell death in MDA-MB-231 cells were assayed using a fluorescence-activated cell sorting (FACS)-based detection kit obtained from Biovision Research Products (Mountain View, CA, USA) [32]. Cells were grown overnight in 6-well plates containing DME/F12 medium supplemented with 10% FBS. Medium was removed, after which cells were washed and then treated for 24 hours with APR-246 or vehicle alone in DME/F12 medium + 5% FBS. Cells were harvested with Accutase and stained with Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI). While Annexin V-FITC reveals the early stages of apoptosis, PI detects DNA fragmentation or cell death. The percentage of Annexin V-FITC-positive/PI-positive cells was determined by measuring the fluorescence of 10,000 cells using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). Experiments were performed at least twice.
Western blotting
Cultured MDA-MB-231 cells were treated with 25 or 50 µM APR-246 in 100-mm culture dishes. After 6-24 hours cells were washed with cold PBS containing phosphatase inhibitors and harvested by gentle scraping with a cell lifter. Whole-cell extracts were prepared using a nuclear extraction TransAm kit (Active Motif). In brief, cells were centrifuged at 200 x g for 5 minutes. Pellets were re-suspended in complete lysis buffer which contained 1 mM dithiothreitol and 1% protease inhibitor cocktail, shaken on ice for 30 minutes, and then centrifuged at 14,000 rpm in an Eppendorf centrifuge at 4°C. Supernatants were aliquoted into microcentrifuge tubes and stored at -80°C. For Western blotting, samples containing 45 µg protein were separated in a NuPAGE 10% Bis-Tris Gel (Invitrogen, Carlsbad, CA, USA). Electrophoresis was performed at 120 V for 1.5 hours in NuPAGE MES-SDS Running Buffer (Invitrogen), after which separated proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA, USA) at 25 V for 25 minutes. Blots were blocked at room temperature for 1 hour with 5% non-fat dry milk in Tris-buffered saline with 0.1% Tween 20 (TBS-T) buffer and then incubated for 2 hours at room temperature with primary antibodies against Bax, p21, or Bcl-2 (1:200 dilution) (Santa Cruz Biotechnology, Dallas, TX, USA) or caspase-3 (1:300 dilution) (R&D Systems, Minneapolis, MN, USA). After being washed three times in TBS-T, blots were incubated with secondary antibody for 1 hour at room temperature, then washed seven times with TBS-T. Immunoreactive bands were visualized using an ECL Plus detection kit (Amersham, Pharmacia Biotech, Arlington Heights, IL, USA). Membranes were stripped and re-probed using β-actin antibody (Sigma-Aldrich) as a control for protein loading.
VEGF ELISA
VEGF levels were measured using the Quantikine human VEGF enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems) [33,34]. MDA-MB-231 cells were grown to 50-60% confluence, washed twice, then treated for 16 hours at 37°C with vehicle or APR-246. Supernatants were collected and the VEGF concentration of each sample measured according to the manufacturer’s protocol. Experiments were performed in quadruplicate, with each sample being analyzed in duplicate on a microplate reader. Inter- and intra-assay coefficients of variance given by the manufacturer for the cell culture supernatant assay were 5-8.5% and 3.5-6.5%, respectively.
Animal studies
The Animal Care and Use Committee at the University of Missouri (Columbia, MO, USA) approved all animal studies. The study followed the guidelines of the U.S. Government Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training. Female athymic nude (nu/nu) mice, 5-6 weeks old and 20-22 g, were purchased from Harlan Sprague Dawley, Inc. (Indianapolis, IN, USA). Mice were housed in a laminar air-flow cabinet under specific pathogen-free conditions. All facilities were approved by the American Association for Accreditation of Laboratory Animal Care in accordance with the current regulations and standards of the U.S. Department of Agriculture, the Department of Health and Human Services, and the National Institutes of Health.
Treatment of nude mice bearing MDA-MB-231 TNBC xenografts with APR-246 and 2aG4
Cultured MDA-MB-231 cells were harvested with trypsin and washed twice with DME/F12 medium. Cells (5 ´ 106) were then re-suspended in 0.15 mL Matrigel (BD Biosciences, Bedford, MA, USA)/DME/F12 medium (4:1 v/v) and injected subcutaneously into both flanks of nude mice. Digital calipers were used every three days to measure tumor size. Tumor volumes were calculated by the formula (L ´ W ´ H) ´ π/6 [35,36]. Once tumor volumes reached around 100-125 mm3, treatment commenced (Day 18). Animals were assigned to one of four groups (Control, APR-246, 2aG4, or APR-246 + 2aG4), with eight mice/group. One group of animals was administered APR-246 alone (100 mg/kg/day) by intravenous (iv) injection into the tail vein. A second group received 2aG4 (100 µg/mouse/day) by intraperitoneal (ip) injection, while a third was given both APR-246 and 2aG4 (100 mg/kg + 100 µg/mouse/day) by iv/ip injection, respectively. Animals were treated every day for a total of 7 treatments, then every other day for additional 11 treatments. One last injection was given 2 hours prior to termination of the experiment. The control group received control antibody C44 (100 µg/mouse/day, ip) and/or PBS (0.1 mL/day, iv). Animal weights were recorded twice weekly throughout the study. At the end of the experiment, mice were sacrificed and tumors harvested. Fresh tumor tissues were immediately placed in 4% paraformaldehyde solution for immunohistochemical analysis or frozen in liquid nitrogen for future studies.
Quantification of immunohistochemical staining
Expression of VEGF in immunohistochemically stained sections of tumor tissue was quantitated by measuring immune-labeled pixels in standardized digital images, photographed at 20X magnification, using the Fovea Pro 3.0 imaging program (Reindeer Graphics, Asheville, NC, USA). VEGF distribution in every cell within each tumor image was determined; 8 to 12 images were analyzed per treatment group from three to five tumors/treatment group. Data are reported as the mean number of labeled pixels per group.
Quantitation of blood vessels was accomplished by photographing tissue sections at 20X magnification from four to five tumors per treatment group that had been stained immunohistochemically for CD31. The total number of vessels in these digital images was counted in 8 to 15 fields per treatment group, with each field representing approximately 0.39 mm2. Vessel density was calculated as vessel number per field.
Statistical analysis
Differences among groups were tested using Student’s t-test or one-way analysis of variance (ANOVA). SigmaPlot software (version 14) was used for statistical analysis. Data are reported as mean ± standard error of the mean (SEM). For all comparisons, P < 0.05 was considered significant. The assumption of the ANOVA was examined, and if necessary, a nonparametric measure based on ranks was used. If normality failed, Kruskal-Wallis one-way ANOVA on ranks was used in place of regular ANOVA. In cases where a significant effect was shown by ANOVA (F-ratio, P < 0.05), the Student-Newman-Keuls multiple comparison test was employed to compare the means of the individual groups.
APR-246 reduces TNBC cell viability in vitro
We first examined the effects of APR-246 on two mtp53-expressing TNBC cell lines: MDA-MB-231 and MDA-MB-468. APR-246 treatment for 24 or 48 hours significantly reduced the viability of both TNBC cell lines, with MDA-MB-468 cells being more sensitive to the drug (Figure 1A). APR-246 had no effect on wtp53-expressing non-TNBC MCF-7 cells until a high (50 µM) cytotoxic concentration of the compound was used. APR-246 exhibited similar effects when used at low concentrations for 5 days (1 nM-5µM). Exposure of TNBC cells to levels of APR-246 as low as 1 nM significantly reduced cell viability, but had no effect on MCF-7 cells (Figure 1B). Importantly, normal mammary cells that express wtp53 (AG11132A) incubated with levels of APR-246 up to 10 µM for 24 hours (which affected TNBC cells significantly) did not reduce their viability (Figure 2), suggesting that toxicity of the compound will be minimal if used clinically.
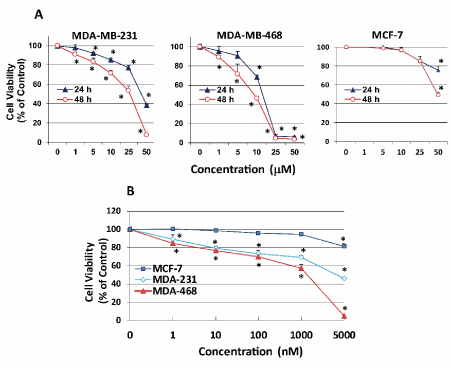
Figure 1. APR-246 reduces cell viability of TNBC cells in vitro. (A) MDA-MB-231 (3.5 ´ 103/well) and MDA-MB-468 TNBC cells (4.5 ´ 103/well) and wtp53-expressing hormone-dependent MCF-7 breast cancer cells (5.5 ´ 103/well) were seeded into a 96-well plate overnight. Cells were washed with culture medium devoid of FBS, then treated for 24 or 48 hours with APR-246 in DME/F12 medium + 5% FBS at the doses shown. (B) MDA-MB-231 cells (2 ´ 104/well), MDA-MB-468 cells (6 ´ 104/well), and MCF-7 cells (5 ´ 104/well) were seeded into 6-well plates overnight, washed with culture medium devoid of FBS, and treated for 5 days with APR-246 in DME/F12 medium + 5% FBS at the doses shown. APR-246 was replaced every 48 hours. Controls were incubated in the absence of APR-246. Cell viability was determined with SRB assays. Data are shown as the mean ± SEM (n=6). *Significantly different from control group; P < 0.05 (ANOVA)
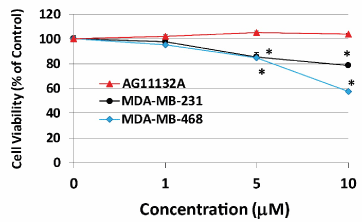
Figure 2. APR-246 does not reduce viability of normal mammary cells in vitro. Normal mammary cells (AG11132A cells) (5 ´ 103/well), MDA-MB-231 TNBC cells (3 ´ 103/well), and MDA-MB-468 TNBC cells (5 ´ 103/well) were seeded into a 96-well plate overnight. Cells were washed with culture medium devoid of FBS, then treated for 24 hours with APR-246 in DME/F12 medium + 5% FBS at the doses shown. Controls were incubated in the absence of APR-246. Cell viability was determined with SRB assays. Data are shown as the mean ± SEM (n=6). *Significantly different from control group; P < 0.05 (ANOVA)
Initial work investigating activators of mtp53 used PRIMA-1, an analogue of APR-246. However, for clinical trials the more permeable analogue, APR-246, has been synthesized and tested [37]. When we compared the potency of both compounds on MDA-MB-231 TNBC cells, APR-246 reduced cell viability significantly more effectively than PRIMA-1 did (Figure 3). After 24 and 48 hours, APR-246 was, respectively, 1.7- and 2.4-fold more potent than PRIMA-1 in reducing MDA-MB-231 cell viability (Table 1).
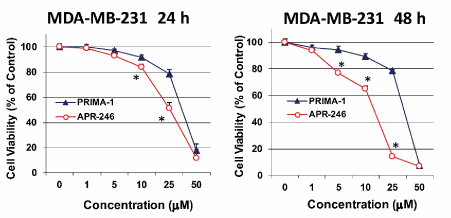
Figure 3. APR-246 reduces MDA-MB-231 TNBC cell viability in vitro more effectively than PRIMA-1. MDA-MB-231 cells (3.5 ´ 103/well) were seeded into a 96-well plate overnight. Cells were washed with culture medium devoid of FBS, then treated for 24 or 48 hours with APR-246 or PRIMA-1 in DME/F12 medium + 5% FBS at the doses shown. Controls were incubated in the absence of APR-246 or PRIMA-1. Cell viability was determined with SRB assays. Data are shown as the mean ± SEM (n=6) *Significantly different from PRIMA-1 group; P < 0.05 (Student’s t-test)
Table 1. IC50 (µM) of PRIMA-1 and APR-246 (MDA-MB-231 cells)
|
PRIMA-1 |
APR-246 |
24 h |
41.65 ± 2.98 |
24.53 ± 0.79 |
48 h |
34.54 ± 0.47 |
14.59 ± 1.72 |
APR-246 activates mtp53 DNA binding in MDA-MB-231 TNBC cells in vitro
Using an oligonucleotide-based DNA-binding assay, we further examined whether APR-246 can convert mtp53 to wtp53 (a form able to bind to a consensus p53 binding site) in MDA-MB-231 cells. APR-246-treated MDA-MB-231 cells exhibited significantly increased DNA binding compared with untreated MDA-MB-231 cells (Figure 4), an effect that occurred rapidly (within 10 minutes) following exposure to APR-246.
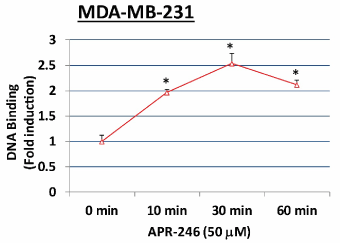
Figure 4. APR-246 activates mtp53 DNA binding in MDA-MB-231 TNBC cells in vitro.
MDA-MB-231 cells were grown in 100-mm culture dishes and treated for 0 (control), 10, 30, or 60 minutes with 50 µM APR-246 in DME/F12 medium + 5% FBS. Activation of p53 in nuclear extracts (2.5 µg) prepared from each group was evaluated by a DNA-binding assay using a TransAM p53 assay kit. Data are shown as the mean ± SEM from three different determinations. *Significantly different from control (0 minutes) group; P < 0.05 (ANOVA)
APR-246 induces apoptosis in MDA-MB-231 TNBC cells in vitro
We next conducted studies to determine whether APR-246 causes apoptosis in mtp53-expressing TNBC cells. Exposure to APR-246 for 24 hours significantly induced apoptosis and cell death in MDA-MB-231 cells in a dose-dependent manner (Figure 5). Analysis of the expression of mitochondria-dependent components of the apoptotic pathway in MDA-MB-231 cells demonstrated that treatment with APR-246 elevated expression of Bax and p21 proteins and caused cleavage of caspase-3 (Figure 6A). Taken together, these studies suggest that APR-246 induces apoptosis in MDA-MB-231 cells by activating a signaling pathway that is mitochondria-dependent. In accord with its ability to induce apoptosis in tumor cells, APR-246 treatment also reduced levels of the survival protein Bcl-2 in MDA-MB-231 cells (Figure 6B).
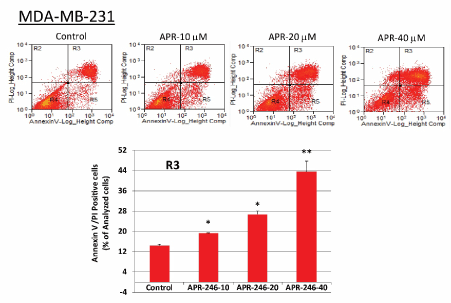
Figure 5. APR-246 induces apoptosis in MDA-MB-231 TNBC cells in vitro. MDA-MB-231 cells were grown in 6-well plates overnight and then treated for 24 hours with APR-246 in DME/F12 medium + 5% FBS at the doses shown. Controls contained the vehicle alone. Apoptosis was measured with a FACS-based apoptosis and cell death assay kit. Representative data are shown; bar graphs represent the percentage of Annexin V-FITC-positive/PI-positive cells (quadrant 3) in the example shown (mean ± SEM from three different determinations). *Significantly different from control group; P < 0.05 (ANOVA). **Significantly different from control, 10, and 20 µM groups; P < 0.05 (ANOVA)
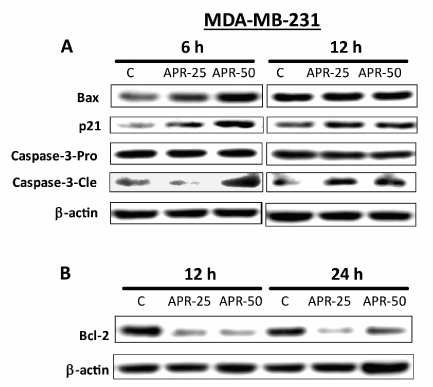
Figure 6. APR-246 increases expression of apoptotic-pathway markers and reduces expression of cell-survival markers in MDA-MB-231 TNBC cells in vitro. (A) MDA-MB-231 cells were grown in 100-mm culture dishes and treated for 6 or 12 hours with 25 or 50 µM APR-246 in DME/F12 medium + 5% FBS. Cells were then washed and harvested. After cell harvest, whole-cell lysates (45 µg protein from each sample) were analyzed by Western blot for Bax, p21, and caspase-3 protein expression. (B) Bcl-2 expression was analyzed under similar conditions as in A, except cells were exposed to APR-246 for 12 or 24 hours. β-actin was used as a loading control. C, control cells treated with vehicle alone
APR-246 alone and in combination with the phosphatidylserine-targeting antibody 2aG4 inhibits growth of MDA-MB-231 TNBC xenografts in nude mice
MDA-MB-231 TNBC tumor xenografts were grown in nude mice. Administration of APR-246 ± 2aG4 monoclonal antibody commenced when tumors were approximately 100-125 mm3 in volume. Tumor volumes and animal weights were monitored for the next few weeks (Figure 7A). Tumors continued to develop in animals administered the control IgG C44 and PBS. However, treatment with APR-246 or 2aG4 alone, or in combination, significantly inhibited tumor growth (Figure 7B). Animals maintained their weight throughout the study (Figure 7C), indicating that the treatment protocol was not toxic. Importantly, a combination of APR-246 and 2aG4 had a synergistic inhibitory effect on tumor growth (Figure 7B) and, over the course of the study, completely eradicated almost 20% of the tumors formed (Figure 8A and 8B) presents examples of tumors in situ at the end of the experiment).
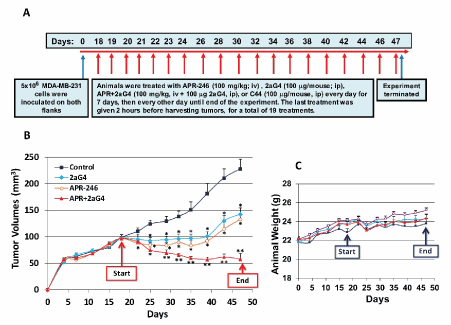
Figure 7. APR-246 alone and in combination with 2aG4 inhibits growth of MDA-MB-231 TNBC xenografts in nude mice. (A) Protocol used for in vivo studies. MDA-MB-231 cells (5 ´ 106) were re-suspended in 0.15 mL Matrigel/DME/F12 medium (without FBS) (4:1 v/v) and used to inoculate nude mice in both flanks. On day 18, animals began to be treated with APR-246, 2aG4, or APR-246 + 2aG4. Control animals were treated with antibody C44. Tumors were measured every 3 days and animals were weighed twice a week. (B, C) Tumor volumes (B) and weights (C) of tumor-bearing animals. Data are shown as the mean ± SEM (n=8). *Significantly different from control; P < 0.05 (ANOVA). **Significantly different from APR-246 alone and 2aG4 alone groups; P < 0.05 (ANOVA)
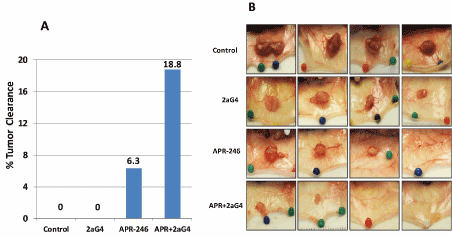
Figure 8 APR-246 and 2aG4 combination therapy eradicates some MDA-MB-231 TNBC tumors in nude mice. Tumors were tracked in the mice inoculated with MDA-MB-231 cells and treated with APR-246, 2aG4, or APR-246 + 2aG4 in the in vivo tumor xenograft experiment described in Figure 7A. (A) The number of tumors formed was determined and monitored throughout the treatment period. At the end of the experiment, the number of tumors that had completely regressed was quantitated, and the percent of tumor clearance for each treatment calculated. (B) Representative images of selected tumors at the end of the treatment regimens used (4 images for each treatment are shown). Complete eradication is demonstrated in the far-right image for APR-246 treatment alone, and the two most right-hand images for APR-246 + 2aG4 treatment
APR-246 and 2aG4 combination treatment reduces VEGF expression and blood-vessel density in MDA-MB-231 TNBC tumor xenografts in nude mice
We next examined tissue sections of tumors obtained from the experiment shown in Figure 7 to ascertain the effects of APR-246 and 2aG4 treatment on tumor blood vessels. APR-246 alone or in combination with 2aG4 significantly reduced blood-vessel density (CD31 expression) in tumor tissue, with the reduction being the most pronounced in the APR-246 + 2aG4 treatment group (Figure 9A). In the same tumor tissue sections, VEGF expression was reduced in tumors from animals given either APR-246 or 2aG4 alone. Combination APR-246 and 2aG4 treatment reduced VEGF expression to a level that was significantly lower than that observed using either agent alone (Figure 9B).
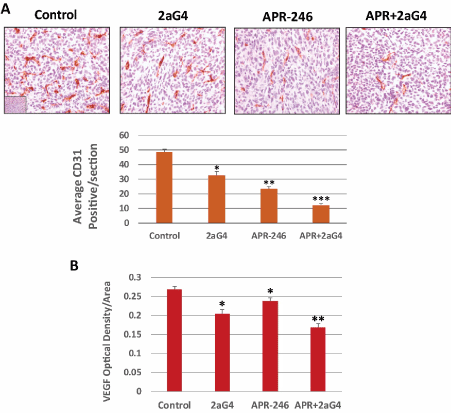
Figure 9 APR-246 and 2aG4 combination therapy reduces angiogenesis markers in MDA-MB-231 TNBC tumor xenografts in nude mice. At the termination of the experiment shown in Figure 7, tumors were harvested and fixed. Tumor tissue sections were then immunohistochemically stained for CD31 or VEGF expression. (A) Upper panel, representative images of CD31 staining in tumor tissue sections. Bar graph, blood vessels were quantitated by photographing CD31-labeled tissue sections from four to five tumors per treatment group at 20X magnification. The total number of vessels in these digital images was counted in 8 to 15 fields per treatment group, with each field representing approximately 0.39 mm2. Vessel density was calculated as vessel number per field. Data are shown as the mean ± SEM (24-25 sections/group from 3-4 animals). *Significantly different from control group; **Significantly different from control and 2aG4 alone groups; ***Significantly different from control, APR-246 alone, and 2aG4 alone groups; P < 0.05 (ANOVA). (B) VEGF expression was quantified using the Fovea Pro 3.0 imaging program. The mean VEGF density was determined from 8–12 images per treatment group (three to five tumors/treatment group). Data are shown as the mean ± SEM (45-60 sections/group from 3-4 animals). *Significantly different from control group; **Significantly different from control, APR-246 alone, and 2aG4 alone groups; P < 0.05 (ANOVA)
APR-246 inhibits VEGF secretion from MDA-MB-231 TNBC cells in vitro
VEGF is a potent angiogenic factor, as well as a survival factor, in breast cancer [38]. For this reason and because we found that APR-246 lowered VEGF expression in xenograft tumors, we performed in vitro studies to determine whether APR-246 also blocks production of VEGF by cultured MDA-MB-231 TNBC cells. Treatment of MDA-MB-231 cells with APR-246 for 24 hours indeed reduced cellular VEGF production (Figure 10).
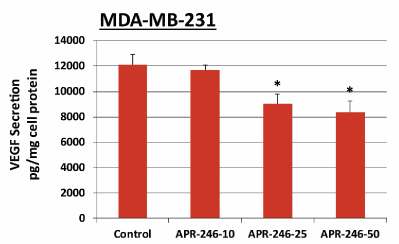
Figure 10. APR-246 inhibits VEGF secretion from MDA-MB-231 TNBC cells in vitro.
MDA-MB-231 cells were grown in 100-mm culture dishes to 50–60% confluence, washed twice with PBS, then treated for 16 hours with 10, 25, or 50 µM APR-246 in DME/F12 medium + 5% FBS. Controls contained the vehicle alone. Culture supernatant (1 mL) was collected from each dish and the VEGF content measured in 200 µL of supernatant using a VEGF ELISA assay kit. Data are shown as the mean ± SEM (n=3). *Significantly different from control group; P < 0.05 (ANOVA)
Previously, we observed in an animal model that treatment with a combination of APR-246 and 2aG4 reduced metastasis of TNBC tumors to the lungs [19]. In the present study, we performed studies to examine the ability of APR-246 to re-activate mtp53 and inhibit the growth of mtp53-expressing TNBC cells and whether APR-246 alone, as well as in combination with a phosphatidylserine-targeting antibody, 2aG4, could inhibit primary TNBC tumor growth.
Functional studies have shown that APR-246 is converted to methylene quinuclidinone, which covalently binds to cysteine residues in the p53 protein, thereby re-activating mtp53 [20,21]. In human clinical trials, doses of up to 135 mg/kg of APR-246 have been administered, with doses between 60-100 mg/kg being well tolerated and found to be clinically useful against hematologic malignancies and prostate cancer [39,40]. However, whether APR-246 might also be used to control TNBC, the most aggressive form of breast cancer, has not been studied in detail.
In previous studies, we showed that PRIMA-1 (APR-246’s parent compound) activated mtp53 in hormone-dependent breast cancer cell lines [41,42]. Here, we confirmed that APR-246 also activates mtp53 in TNBC cells. In addition, both APR-246 and PRIMA-1 reduced the viability of TNBC cells in vitro, though APR-246, the methylated and more permeable form of PRIMA-1, was slightly more potent.
Our in vivo analysis using a tumor xenograft mouse model showed that APR-246, either alone, or administered in combination with 2aG4, very effectively inhibited the growth of TNBC tumors. Combination therapy was most effective in preventing the growth of tumors in the timeframe tested, and completely eliminated a number of tumors present prior to treatment. This result is most likely due to APR-246-induced apoptosis, followed by disruption and dissolution of blood vessels by 2aG4, given that tumor sections from treated groups showed a loss of blood vessel density, as well as significantly reduced expression of the potent angiogenesis factor VEGF.
A number of our findings suggest that APR-246 could be safe and effective in clinical use. First, the use of APR-246 concentrations in the nM range for 5 days effectively reduced the viability of TNBC cells in vitro, indicating that long-term use of low concentrations of this agent will likely be effective in humans. Second, APR-246 had no effect on normal AG11132A mammary cells, suggesting that it could be used in vivo with little to no toxicity to normal tissues. Third, the animals in our in vivo experiments maintained their weight throughout the study, suggesting that our treatment protocols appear to be non-toxic. Indeed, clinical trials involving patients with hematological and prostate cancers suggest that APR-246 is relatively safe for human use.
We showed that APR-246 targets and activates mtp53 in TNBC cells in vitro. Furthermore, when administered alone or in combination with 2aG4, APR-246 reduced VEGF expression, disrupted angiogenesis, and effectively inhibited the growth of TNBC tumor xenografts in vivo. Because almost 80% of TNBCs express mtp53, we contend that APR-246 should be tested for its efficacy against TNBC in clinical trials. Our results show that APR-246 was particularly effective when administered with 2aG4, which disrupts tumor blood vessels and may also activate an immune response in tissues [43]. We therefore propose that APR-246 + 2aG4 therapy could benefit women afflicted with TNBC, for whom there are currently few effective non-toxic treatment options. Furthermore, since p53 mutations are common in a variety of cancers, and angiogenesis is an essential component of metastasis in all tumors [44-46], we contend that our findings justify the future study of APR-246 and 2aG4 for treating a variety of human cancers other than TNBC [45,46].
Financial support was provided by a peer-reviewed faculty research grant from the College of Veterinary Medicine, University of Missouri and in part by APREA AB. APREA AB provided the APR-246 that was used in the in vivo experiments without charge. However, APREA AB did not influence the experiments conducted or the reporting of results. SMH is the Zalk Missouri Professor of Tumor Angiogenesis.
The authors report no conflicts of interest in this work.
- Siegel RL, Miller KD, Jemal A (2020) Cancer statistics, 2020. CA Cancer J Clin 70: 7-30.
- Garrido-Castro AC, Lin NU, Polyak K (2019) Insights into molecular classifications of triple-negative breast cancer: Improving patient selection for treatment. Cancer Discov 9: 176-198. [Crossref]
- Lyons TG (2019) Targeted therapies for triple-negative breast cancer. Curr Treat Options Oncol 20: 82.
- da Silva JL, Cardoso Nunes NC, Izetti P, de Mesquita GG, de Melo AC (2020) Triple negative breast cancer: A thorough review of biomarkers. Crit Rev Oncol Hematol 145: 102855.
- Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490: 61-70.
- Walerych D, Napoli M, Collavin L, Del Sal G (2012) The rebel angel: mutant p53 as the driving oncogene in breast cancer. Carcinogenesis 33: 2007-2017. [Crossref]
- Dhakal HP, Naume B, Synnestvedt M, Borgen E, Kaaresen R, et al. (2012) Expression of vascular endothelial growth factor and vascular endothelial growth factor receptors 1 and 2 in invasive breast carcinoma: prognostic significance and relationship with markers for aggressiveness. Histopathology 61: 350-364.
- Berns EM, Klijn JG, Look MP, Grebenchtchikov N, Vossen R (2003) Combined vascular endothelial growth factor and TP53 status predicts poor response to tamoxifen therapy in estrogen receptor-positive advanced breast cancer. Clin Cancer Res 9: 1253-1258.
- Nishizaki M, Fujiwara T, Tanida T, Hizuta A, Nishimori H (1995) Recombinant adenovirus expressing wild-type p53 is antiangiogenic: a proposed mechanism for bystander effect. Clin Cancer Res 5: 1015-1023. [Crossref]
- André F, Job B, Dessen P, Tordai A, Michiels S, Liedtke C (2009) Molecular characterization of breast cancer with high-resolution oligonucleotide comparative genomic hybridization array. Clin Cancer Res 15: 441-451.
- Turner N, Moretti E, Siclari O, Migliaccio I, Santarpia L, et al. (2013) Targeting triple negative breast cancer: is p53 the answer? Cancer Treat Rev 39: 541-550.
- Mohammed RA, Ellis IO, Mahmmod AM, Hawkes EC, Green AR (2011)Martin SG Lymphatic and blood vessels in basal and triple-negative breast cancers: characteristics and prognostic significance. Mod Pathol 24: 774-785.
- Bassett EA, Wang W, Rastinejad F (2008) Structural and functional basis for therapeutic modulation of p53 signaling. Clin Cancer Res 14: 6376-6386.
- Rivlin N, Brosh R, Oren M, Rotter V (2011) Mutations in the p53 Tumor Suppressor Gene: Important milestones at the various steps of Tumorigenesis. Genes Cancer 2: 466-474.
- Rahko E, Blanco G, Soini Y, Bloigu R, Jukkola A (2003) A mutant TP53 gene status is associated with a poor prognosis and anthracycline- resistance in breast cancer patients. Eur J Cancer 39: 447-453.
- Spike BT, Wahl GM (2011) p53, stem cells, and reprogramming: Tumor suppression beyond guarding the genome. Genes Cancer 2: 404-419.
- Godar S, Ince TA, Bell GW, Feldser D, Donaher JL (2008) Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell 134: 62-73.
- Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y (2011) p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol 13: 317-323.
- Liang Y, Besch-Williford C, Cook MT, Belenchia A, Brekken RA (2019) APR-246 alone and in combination with a phosphatidylserine-targeting antibody inhibits lung metastasis of human triple-negative breast cancer cells in nude mice. Breast Cancer: Targets and Therapy 11: 249-259.
- Kaar JL, Basse N, Joerger AC, Stephens E, Rutherford TJ (2010) Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci 19: 2267-2278.
- Lambert JM, Gorzov P, Veprintsev DB, Söderqvist M, Segerbäck D (2009) PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 15: 376-388.
- Zandi R, Selivanova G, Christensen CL, Gerds TA, Willumsen BM (2011) PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin Cancer Res 17: 2830-2841. [Crossref]
- Synnott NC, Murray A, McGowan PM, Kiely M, Kiely PA (2017) Mutant p53: a novel target for the treatment of patients with triple-negative breast cancer? Int J Cancer 140: 234-246.
- Ran S, He J, Huang X, Soares M, Scothorn D (2005) Antitumor effects of a monoclonal antibody that binds anionic phospholipids on the surface of tumor blood vessels in mice. Clin Cancer Res 15: 1551-1562.
- He J, Luster TA, Thorpe PE (2007) Radiation-enhanced vascular targeting of human lung cancers in mice with a monoclonal antibody that binds anionic phospholipids. Clin Cancer Res 13: 5211-5218.
- He J, Yin Y, Luster TA, Watkins L, Thorpe PE (2009) Antiphosphatidylserine antibody combined with irradiation damages tumor blood vessels and induces tumor immunity in a rat model of glioblastoma. Clin Cancer Res 15:6 871-6880.
- Luster TA, He J, Huang X, Maiti SN, Schoroit AJ (2006) Plasma protein beta-2-glycoprotein 1 mediates interaction between the anti- tumor monoclonal antibody 3G4 and anionic phospholipids on endothelial cells. J Biol Chem 281: 29863-29871.
- Huang X, Bennett M, Thorpe PE (2005) A monoclonal antibody that binds anionic phospholipids on tumor blood vessels enhances the antitumor effect of docetaxel on human breast tumor in mice. Cancer Res 65: 4408-4416.
- Liang Y, Wu J, Stancel GM, Hyder SM (2005) P53-dependent inhibition of progestin-induced VEGF expression in human breast cancer cells. J Steroid Biochem Mol Bio 93: 173-182.
- Rubinstein LV, Shoemaker RH, Paull KD, Simon RM, Tosini S (1990) Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl Cancer Inst 82: 1113-1118.
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J (1990) New colorimetric cytotoxicity assay for anti-cancer-drug screening. J Natl Cancer Inst 82: 1107-1112.
- Cook MT, Liang Y, Besch-Williford C, Hyder SM (2016) Luteolin inhibits lung metastasis, cell migration, and viability of triple-negative breast cancer cells. Breast Cancer: Target and Therapy 9: 9-19.
- Carroll CE, Liang Y, Benakanakere I, Besch-Williford C, Hyder SM (2013) The anticancer agent YC-1 suppresses progestin-stimulated VEGF in breast cancer cells and arrests breast tumor development. Int J Oncol 42: 179-187.
- Liang Y, Hyder SM (2005) Proliferation of endothelial and tumor epithelial cells by progestin-induced vascular endothelial growth factor from human breast cancer cells: paracrine and autocrine effects. Endocrinology 146: 3632-3641.
- Liang Y, Mafuvadze B, Besch-Williford C, Hyder SM (2018) A combination of p53-activating APR-246 and phosphatidylserine-targeting antibody potently inhibits tumor development in hormone-dependent mutant p53-expressing breast cancer xenografts. Breast Cancer: Target and Therapy 10: 53-67.
- Liang Y, Besch-Williford C, Hyder SM (2009) PRIMA-1 inhibits growth of breast cancer cells by re activating mutant p53 protein. Int J Oncol 35: 1015-1023.
- Perdrix A, Najem A, Saussez S, Awada A, Journe F (2017) PRIMA-1 and PRIMA-1Met (APR-246): from mutant/wild type p53 reactivation to unexpected mechanisms underlying their potent anti-tumor effect in combinatorial therapies. Cancers 9: 172. [Crossref]
- Liang Y, Brekken RA, Hyder SM (2006) Vascular endothelial growth factor induces proliferation of breast cancer cells and inhibits the anti-proliferative activity of anti-hormones. Endocr Relat Cancer 13: 905-919.
- Lehmann S, Bykov VJ, Ali D, Andrén O, Cherif H (2012) Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol 30: 3633-3639. [Crossref]
- Deneberg S, Cherif H, Lazarevic V, Andersson PO, von Euler M (2016) An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer 6: e447.
- Liang Y, Besch-Williford C, Brekken RA, Hyder SM (2007) Progestin-dependent progression of human breast tumor xenografts: a novel model for evaluating antitumor therapeutics. Cancer Res 67: 9929-9936.
- Liang Y, Besch-Williford C, Benakanakere I, Hyder SM (2007) Re-activation of the p53 pathway inhibits in vivo and in vitro growth of hormone-dependent human breast cancer cells. Int J Oncol 31: 777-784.
- Yin Y, Huang X, Lynn KD, Thorpe PE (2013) Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol Res 1: 256-268.
- Denekamp J (1993) Angiogenesis, neovascular proliferation and vascular pathophysiology as targets for cancer therapy. Br J Radiol 66: 181-196.
- Cheng X, Li L, Thorpe PE, Yopp AC, Brekken RA (2016) Antibody-Mediated blockade of phosphatidylserine enhances the antitumor effect of sorafenib in hepatocellular carcinomas xenografts. Ann Surg Oncol 23: 583-591.
- Burrows FJ, Thorpe PE (2004) Vascular targeting agents as cancer therapeutics. Clin Cancer Res 10: 415-427.