Since the discovery of tyrosine kinase inhibitors in treatment of lung cancer harboring such actionable targets, many lives have been prolonged. To the same extent, same group of patients have failed to benefit from this category of drugs, in long run, either initially or during the course of treatments, simply due to either known or unknown mechanism of resistance which occurs very often in the first few months after initiation of therapy. The resistance is 100 percent expected, and no patient is reported to be a waiver of such pattern. With best practices of oncology, the average duration of response is expected to be below 12 months. About half of these resistance is caused by mutation at T790M in EGFR target, which can be revealed by liquid biopsy. The most recent studies have revealed the significant role of epigenome in controlling this complicated resistance pattern. We have learned that Histone deacetylation, as opposed to promoter methylation, may contribute to the epigenetic silencing and to EGFR TKI resistance in NSCLC. Here we present a model of combinational therapy that targets the EGFR molecule, (by small molecule inhibitor, Afatanib) with simultaneous epigenetic modification of the target, (by application of multitargeted epigenetic therapy (MTET) with significantly improved clinical results. We propose further trials are needed to support such hypothesis, which if proved, could significantly shift the current practices in management of this set of cases in lung adenocarcinomas.
epidermal growth factor, lung cancer, epigenetic therapies, targeted therapies, afatinib
Several categories of resistance have been speculated to be responsible for molecular resistance in EGFR mutated and treated cases with EGFR inhibitors. Most common one is the Gatekeeper mutation in EGFR: T790M mutation. Although most common, this has better post progression/survival prognosis (PPS) compared to other groups of patients with other mechanisms of resistance. Approximately 50% of the acquired resistance developed to erlotinib or gefitinib is linked to T790M mutation and the proportion could be underestimated as more accurate prevalence of 68% was achieved using LNA-PCR/sequencing assay. Second is Compensatory contribution of other RTKs: MET receptor, a trans-membrane tyrosine kinase encoded by proto-oncogene MET, has been highlighted as an important cause for acquired resistance of NSCLC to gefitinib or erlotinib [1-5]. Due to connection of Hepatocyte growth factors to Met pathway, it is speculated that in about 61 percent of cases, studies in Japan, HGF overexpression was responsible in promoting drug resistance. This mechanism is INDEPENDENT of TKI pathways [6]. Third mechanism of resistance is Activation of compensatory signaling pathways, mostly reported Pi3k/ Akt/ M-tor pathway. AKT activation and mTOR phosphorylation were frequently present in NSCLC patients (43-90% and 60-90%, respectively. That said application of several dual targeted therapies to target Pi3k/Akt, has not resulted in improved survival in these patients. Her 2 alteration is seen in about 2 percent of cases [7]. Finally, Epidermo mesenchymal Transition (EMT) phenotypic transformation has been proven in at least 5 percent of cases with EGFR resistance as main mechanism [8-10].
Since Met/ PI3K and EMT transition are all targets for epigenetic modification it appears reasonable for addition of histone deacetylase inhibitors in combination of DNA demethylating agents, in a package under multitargeted Epigenetic Therapy protocol (MTET) to the patients to both prevent and treat the molecular resistance.
What to our knowledge was never reported was addition of epigenetic therapies in clinic to patients with EGFR mutation carcinomas, specifically in case of T790 M mutation. Here we study a case series of 2 patients with such phenomenon and report a significant effect size in their response when epigenetic therapies are implemented under the multitargeted Epigenetic Therapy protocol (MTET)
2 cases with advanced stage four lung adenocarcinoma were treated with combination of a histone deacetylase inhibitor (PB) and a polyphenol known for DNA demethylating effect. The therapy was provided through intravenous administration. The patients were counselled about their care and consented appropriately. Early molecular response was defined by measurable change in the MAF after first cycle of therapy in two weeks. Major molecular response was defined by 2.5 log decrease in mutated allele fraction (MAF) of altered genes. The response met the criteria for early and major molecular response (EMR/ MMR).
Case 1
54 years old female with history of EGFR mutated lung adenocarcinoma, metastatic to brain, status post gamma knife on 10/16, and left cerebellar resection on 10/16 and left VATS procedure on 4/2017, Gliotrif started in 2016, with good response for about 3 months, further progression of her disease in her skeletal bones and lungs, negative T790M mutation in her Travogene test, referred to us in February 2018, for evaluation and treatments.
At the time of her initial evaluation, she had pain in her back, as a result of her tumor in her spine (T4), she also had noticed an enlarged left axillary node, for about 2 months, Her CEA had increased from 11 to 26, in 6 months.
Our initial labs confirmed the presence of EGFR mutation at T790 M, as well as increased CYFRA 21.1 and IGF-1. Immediately she was started on IV epigenetic therapies per MTET protocol, which she received on daily basis for two weeks. Her labs further were repeated on March 9th, which showed reduction of her circulating DNA MAF (mutation allele fraction) from 31 percent down to 5 percent in her EGFR. (plus, APC, Kit, BRAF, MET alteration reduced or disappeared). Her blood test also confirmed reduction of her ALK-P, CRP and LDH. (normalized) Her IGF-1 also normalized at 118 (on 3/9/18) (Figure 1).
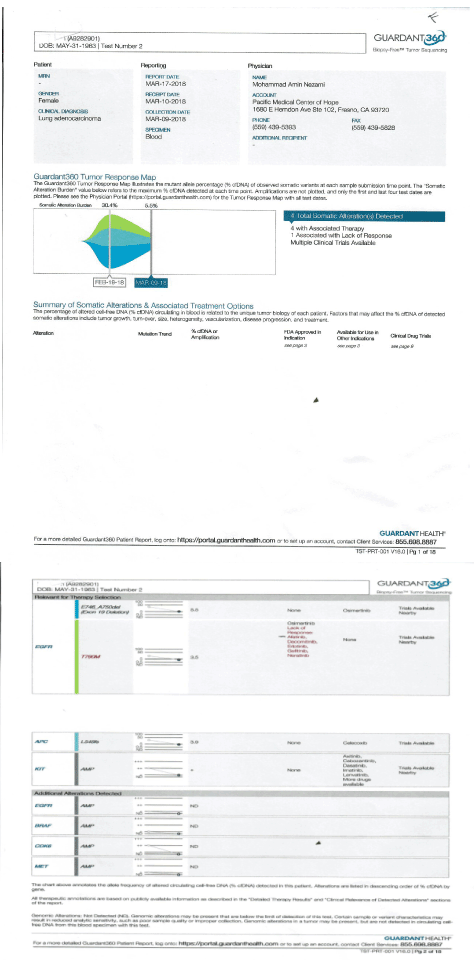
Figure 1. Circulating DNA, Pre and Post MTET therapy.
Case 2
55 years old male with history of right upper lobe mass in the lung about 4.5 cm in size diagnosed as adenocarcinoma, EGFR positive (he had been diagnosed after a LN biopsy, which was positive after a period of time when he was only suspected to have unknown primary adenocarcinoma in October 2017). He unfortunately had wide spread bone (sternum, spine, femur, pelvic) and brain lesions (frontoparietal lobe and cerebellum), was unable to walk, due to wide metastatic disease in brain and cerebellum, further he was treated with cyberknife and Afatanib, since December 2017, as his tumor was EGFR positive. He has been referred to us by his PMD for evaluation and treatments.
His bone scan had shown several areas of metastatic disease, including bilateral ribs, sacrum, cervical, thoracolumbar spine, left humerus, femur, ilia bone, and sternum, on 10/29/17. His CT of chest showed innumerous pulmonary nodules, as well as a large mass in right upper lobe, in the size of 4.5 cm X 2.5 cm, as well as lesions in left lung apex and pleural based nodules, multiple mediastinal and hilar LNs. His MRI of brain had shown, multiple lesions, largest in right frontoparietal lobe with 2.3×2.3 cm size, along with second largest in midline, 1.5 cm in size, many smaller lesions, with accompanying hemorrhagic features and vasogenic edema. CT of abdomen, multiple liver lesions identified.
Upon his arrival, his labs were drawn, and it showed increased CEA at 14.4. His liquid biopsy was positive for RB1, and EGFR, collected on 12/11/17 (Figure 2).
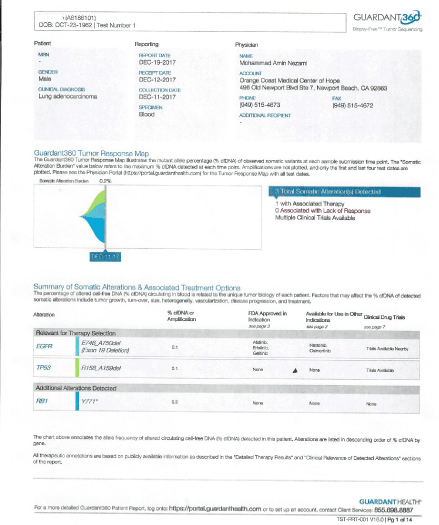
Figure 2. Circulating DNA Pre treatment.
Immediately he was started on IV epigenetic therapies which he received on daily basis, starting in December 5th, 2017. Immediately he was feeling better, and able to walk after two weeks of therapy.
His post treatment labs after ten days showed CEA decreased from 14.4, measured on 12/8/17 down to 10.6 measured on 12/21/17. Further his TGF and CEA dropped again after two weeks of therapies. His TGF dropped down to 9235 from 12201, and CEA decreased to 5.1 on 1/11/8. Further tested on 1/18/18 and it came down to 3.7 (normalized). Clinically he improved, with ability to stop the steroids all together.
His c DNA showed a drastic response after two weeks of treatments, with RB1 and TP53 both non-detectable (Figure 3).

Figure 3. cDNA post MTET therapy
This was repeated on 3/16/18 and still was non-detectable. At this time patient was receiving once a week therapy. His oncologist ha called his progress "exceptional". His radiological response has confirmed his skeletal lesions essentially resolved on 2/20/18 scan and his brain lesions have also dropped from 2.3 cm largest mass to 9×6 mm in size verified in his MRI of 2/20/18.
Further his TGF dropped down to 8884 from 12202 in 8 weeks. (measured on 2/2/18 and 12/2/17). His Brain MRI showed 70 percent reduction in all brain lesions volumetric size in February 2018. On 4/9/18 his MRI showed that there was reduction of brain mets in right parietal lobe from 9 to 6 mm, and this time no enhancement seen, Left parietal focus down to 2 mm from 4. Previous left posterior frontal, right inferior frontal, right cerebellar, foci no longer seen.
His PET scan on 2/20/18 showed complete resolution of wide metastatic disease in the skeletal bone as well as pleura, abdomen, pelvis, right hilar, LN. Her pulmonary mass also appeared smaller with less peri bronchovascular changes. The size decreased from 2.9 cm to 0.9 cm and the SUV activity dropped from 9.1 to 1.3.
Then between months of 6/13/18 and 8/19/18 he was unable to receive epigenetic treatments and continued afatinib alone. As a result, patient developed new lesion in his brain MRI along with increased circulating DNA (evident in his liquid biopsy) dated August 9th, 2018 (Figure 4).
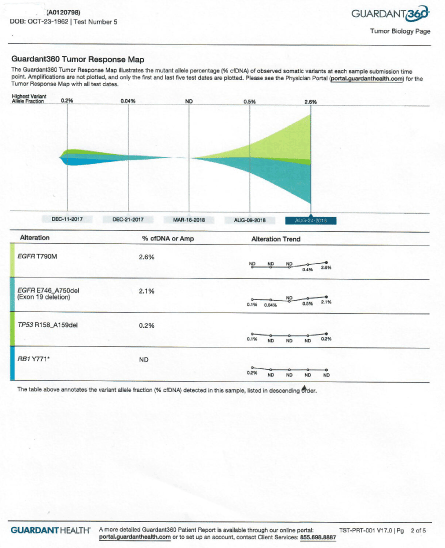
Figure 4. Circulating DNA after withdrawal of MTET therapy
In case number 1, a mutated EGFR tumor responds to epigenetic therapy in combination with afatinib. (where afatanib by itself had failed).
In case number 2, a non-mutated EGFR responds to Afatinib in combination with epigenetic therapy in an expedited fashion, and discontinuation of epigenetic therapy, causes development of resistance to Afatinib through mutation of T790M.
Since the only variable during this time was discontinuation of Epigenetic therapies, and the relapses were secondary to development of EGFR T790 M mutation, we conclude that epigenetic therapies had caused durable response in combination with EGFR inhibition both in non-mutated and mutated EGFR target at T790M.
Treatment of EGFR mutated adenocarcinoma is complex and prolonged survival is challenging. This study although represents small number of cases, but the effect size is major in both cases, apparent on major molecular response reported. We recommend a controlled trial with combination of MTET and EGFR targeted therapy to prove such concept with hypothesis of accomplished durable response as primary aim. We also propose implementation of liquid biopsy to monitor response in these subset of patients as early as days after initiation of epigenetic therapy. We believe such results could impact the standard of care in treating lung cancer and providing a meaningful improved survival to the patients with advanced disease.
- Zhao M, Zhang Y, Li J, Li X, Cheng N, et al. (2018) Histone deacetylation, as opposed to promoter methylation, results in epigenetic BIM silencing and resistance to EGFR TKI in NSCLC. Oncol Lett 15: 1089-1096. [Crossref]
- Sullivan I, Planchard D (2016) Next-generation EGFR tyrosine kinase inhibitors for treating EGFR-mutant. Front Med (Lausanne) 3: 76. [Crossref]
- Sakr L, Kasymjanova G, Small DI, Cohen V, Galvis L, et al. (2017) Lung cancer-non-small cell metastatic 34: 15, predicting duration of response to EGFR TKI among EGFR-mutant NSCLC patients. Journal of Clinical Oncology.
- Kwapisz D (2017) The first liquid biopsy test approved. Is it a new era of mutation testing for non-small cell lung cancer? Ann Transl Med 5: 46. [Crossref]
- Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, et al. (2011) Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res 17: 1169-1180. [Crossref]
- Yano S, Yamada T, Takeuchi S, Tachibana K, Minami Y, et al. (2011) Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol 6: 2011-2017. [Crossref]
- Britten CD, Adjei AA, Millham R, Houk BE, Borzillo G, et al. (2014) Phase I study of PF-04691502, a small-molecule, oral, dual inhibitor of PI3K and mTOR, in patients with advanced cancer. Invest New Drugs 32: 510-517. [Crossref]
- Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, et al. (2005) Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res 11: 8686-8698. [Crossref]
- Thomson S, Buck E, Petti F, Griffin G, Brown E, et al. (2005) Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res 65: 9455-9462. [Crossref]
- Lin Y, Wang X, Jin H (2014) EGFR-TKI resistance in NSCLC patients: mechanisms and strategies. Am J Cancer Res 4: 411-435. [Crossref]