Abstract
We investigated the neuroprotective potential of the peptide mixture cerebrolysin on the development of brain oxidative stress and neuronal injury in rats intoxicated with malathion. Rats were treated with malathion (150 mg/kg, intraperitoneally; i.p.) alone or combined with cerebrolysin at doses of 24.5, 43 or 86 mg/kg or atropine (1 mg/kg) combined with cerebrolysin at doses of 24.5 or 86 mg/kg. Rats were euthanized four hours later, and their brains were analyzed for biochemical indices of oxidative stress, paraoxonase-1 (PON-1), acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) activities. Histopatholoical examination of the brain tissue was also performed. Malathion caused significant increase in brain lipid peroxidation (malondialdehyde) and nitric oxide levels along with a significant decrease in the level of reduced glutathione. Brain AChE, BChE and PON-1 activities were markedly inhibited by malathion exposure. Cerebrolyain given at the above doses resulted in a significant and dose-dependent decrease in malondialdehyde and nitric oxide levels. The drug also almost restored reduced glutathione level, AChE and PON-1 activities to their saline control values. Neuronal degeneration, gliosis, and eosinophilic plaques were observed in cerebral cortex of malathion treated rats. These pathological changes were ameliorated by concomitant treatment with cerebrolysin alone or in combination with atropine. These data provide evidence that cerebrolysin could protect against malalthion neurotoxicity by attenuating oxidative stress.
Key–words
Cerebrolysin; malathion; oxidative stress; acetylcholinesterase; neurotoxicity
Introduction
Neurotoxicity caused by organophosphate insecticides in a recognized health problem [1]. These agents are widely used in agriculture, veterinary and in the household [2]. Acute toxicity is largely due to excessive cholinergic activity [3]. Organophosphates bind to and inhibit the cholinesterases; acetylcholinesterase (EC 3.1.1.7; AChE) and butyrylcholinesterase (EC 3.1.1.8’ BChE). In the central and peripheral nervous system, acetylcholinesterase (AChE) is the enzyme that hydrolyzes the neurotransmitter acetylcholine (ACh), thereby, terminating its action at the cholinergic synapse [4]. Organophosphates thus cause the accumulation of ACh in the synaptic cleft and over stimulation of the cholinergic receptors at post-synaptic cells and/or end organs, and the development of signs of acute toxicity such as excessive salivation, lacrimation, bradycardia, tremors, convulsions, and ultimately respiratory depression and coma [3-6]. Long term exposure to organophosphate insecticides is also associated with central and peripheral neurotoxicity in the form of cognitive and memory impairments, ataxia, delayed peripheral neuropathy [7-9]. Organophosphates have also been incriminated in the development of neurodegenerative diseases like Parkinson’s disease [10-12]. In this context, individuals with inefficient catalytic activity of paraoxonase 1, an enzyme important for the detoxification of organophosphates are more prone to develop Parkinson’s disease upon exposure to these insecticides [12-14].
While inhibition of AChE activity is considered the primary mechanism accounting for neurotoxicty due to organophosphates, other mechanisms such as oxidative stress [15,16], impairment of mitochondrial dynamics [17] and mitochondrial bioenergitics [18,19] are increasingly being implicated. Oxidative stress is the term used to describe the situation where there is an increased generation of reactive oxygen metabolites so as to overcome the cell’s antioxidant mechanisms with ensuing threat to cellular integrity or actual damage to its macromolecules eg., cell membrane lipids, proteins and DNA [20]. Oxidative stress is particularly important in the development of brain neurodegeneration in view of the high rate of oxygen utilization, the auto-oxidation of monoamine neurotransmitters, the presence of redox-active metals, and the rich content of polyunsaturated fatty acids, the preferred target for reactive oxygen metabolites. The brain tissue in addition is deficient in antioxidants compared with other organs [21,22].
In the treatment of acute toxicity caused by organophosphate insecticide, the muscarinic receptor antagonist atropine is the antidote most commonly employed. Meanwhile, cholinesterase reactivators eg., pralidoxime and obidoxime are used to reverse neuromuscular blockade [23,24]. The benefit from oximes is, however, not clear [25]. There is thus a clear need for finding new therapeutic drug options for treating organophosphate neurotoxicty.
Cerebrolysin is a peptide preparation derived from purified porcine brain proteins and consisting of a mixture of low molecular weight peptides and free amino acids. The effect of the drug is thus similar to that of endogenous neurotrophic factors promoting brain protection and repair [26]. The drug has been shown to improve neuropsychiatric manifestations and cognitive function in patients with Alzheimer’s disease and dementia of vascular origin [27], and to improve neurologic outcome in vascular dementia [28] and in patients with acute stroke [29]. Cerebrolysin exerts important pharmacological actions including increased synaptic regeneration [30], enhanced neurogenesis [31], decreased apoptosis [32], increased dendritic spine density and dendritic length [33] as well as antioxidative and anti-inflammatory effects [34,35].
The aim of the present study is therefore to investigate the effect of treatment with cerebrolysin on neuronal injury and oxidative/nitrosative stress evoked in rat brain by the organophosphorus compound malathion.
Materials and methods
Animals
Male Sprague-Dawley rats, weighing 160–180 g of body weight was used in the study. Rats were group housed under temperature- and light-controlled conditions and provided with standard laboratory food and water ad libitum. Animal procedures were performed in accordance with the Ethics Committee of the National Research Centre and followed the recommendations of the National Institutes of Health Guide for Care and Use of Laboratory Animals (Publication No. 85-23, revised 1985).
Drugs and Chemicals
Malathion (Commercial grade, 57%, Naser Chemical Co., A.R.E), cerebrolysin (EVER Neuro Pharma, GmbH, Unterach, Austria)), and atropine (Sigma, St Louis, MO, USA) were used in the experiments. Other chemicals and reagents were purchased from Sigma, USA. Drugs were dissolved in isotonic (0.9%, NaCl) saline solution immediately before use. The doses of cerebrolysin were based on previous studies.
Study Design
Rats were randomly allocated into seven equal groups, six rats each. Group 1 (normal control) received 0.9% saline intraperitoneally (i.p.) (0.2 ml/rat). Group 2–7 received malathion (150 mg/kg, i.p.). Along with malathion injection, groups 3–5 were treated with i.p. cerebrolysin at doses of 21.5, 43 or 86 mg/kg. Group 6 received a combination of cerebrolysin at dose of 21.5 mg/kg and atropine at dose of 1 mg/kg, i.p. Group 7 received a combination of cerebrolysin at dose of 86 mg/kg and atropine at dose of 1 mg/kg, i.p. Rats were by euthanized 4 h later decapitation for tissue collection, their brains then quickly dissected out on an ice-cold plate, washed with ice-cold phosphate-buffered saline (pH 7.4), weighed, and stored at −80 for the biochemical assays. The brain was homogenized with 0.1 M phosphate buffer saline at pH 7.4 to give final concentration of 20% w/v and homogenates were used in biochemical assays.
Biochemical analyses
Determination of lipid peroxidation
The lipid peroxidation was determined by measuring thiobarbituric acid reactive species (TBARS) and the produced red color was then red at 532 nm using spectrophotometer (Shimadzu, Japan) [36].
Determination of nitric oxide
Nitric oxide was determined using Griess reagent. Nitrate is converted to nitrite via nitrate reductase. Griess reagent then act to convert nitrite to a deep purple azo compound and the absorbance was read at 540 nm using spectrophotometer [37].
Determination of reduced glutathione
The content of reduced glutathione was determined according to the method of Ellman, et al. [38]. DTNB (5, 5’-dithiobis (2-nitrobenzoic acid) (Ellman´s reagent) is reduced by the free SH group on GSH molecule and the yellow colored 5-thio-2-nitrobenzoic acid produced is read spectrophotometrically at 412 nm.
Determination of paraoxonase 1 Activity
Paraoxonase-1 arylesterase activity in brain and liver homogenates was determined with the use of phenylacetate as a substrate. Paraoxonase-1 catalyzes the cleavage of phenyl acetate with the formation of phenol. The rate in which phenol is produced is measured using spectrophotometer by monitoring the increase in absorbance at 270 nm and 25°C. One unit of arylesterase activity is defined as 1 μM of phenol formed per minute. Enzyme activity is calculated based on the extinction coefficient of phenol of 1310 M-1 cm-1 at 270 nm, pH 8.0 and 25°C. Enzyme activity is expressed in kilo International Units/Liter (kU/L) [39,40].
Determination of acetylcholinesterase activity
Acetylcholinesterase activity was measured using the method of Ellman, et al. [41,42]. The assay uses DTNB to measure the amount of thiocholine produced as acetylthiocholine is hydrolyzed by AChE. The color of DTNB adduct is measured using spectrophotometric at 412 nm. AChE activity was expressed as μmol sulfhydryl (SH) groups/g. tissue/min.
Determination of butyrylcholinesterase activity
Butyrylcholinesterase activity was measured using commercially available kit from Ben Biochemical Enterprise (Milan, Italy). BChE catalyzes the hydrolysis of butyrylthiocholine as substrate into butyrate and thiocholine. Thiocholine then reacts with 5, 5’-dithiobis (2-nitrobenzoic acid) (DTNB) forming a yellow chromophore which then can be quantified with spectrophotometer [41].
Histopathological examination
The brain of different groups was removed and fixed in 10% formol saline. Paraffin sections (5 μm thick) were stained with haematoxylin and eosin (Hx & E) and investigated by light microscope using digital camera (DP70, Tokyo).
Statistical analysis
Data were expressed as mean ± SEM values. Statistical analysis was performed using one-way ANOVA followed by Duncan post hoc test for intergroup comparison.
Results
- Biochemical results
- Oxidative stress
- Lipid peroxidation
Rats treated with malathion exhibited significantly increased lipid peroxidation, as assessed by malondialdehyde (MDA) levels (49.3% increase; 30.93 ± 1.66 vs. 20.72 ± 1.5 nmol/g. tissue). Cerebrolysin given at 43 or 86.1 mg/kg resulted in a significant decrease in MDA by 18.4-29.1% (25.2 ± 1.36 and 21.9 ± 1.0 vs. 30.93 ± 1.66 nmol/g.tissue). Rats treated with cerebrolysin at 21.5 or 86.1 mg/kg together with atropine resulted in 17.2% and 24.3% decrements in MDA level, respectively, with respect to malathion control value (25.6 ± 1.4 and 23.4 ± 1.2 vs. 30.93 ± 1.66 nmol/g.tissue) (Figure 1A).
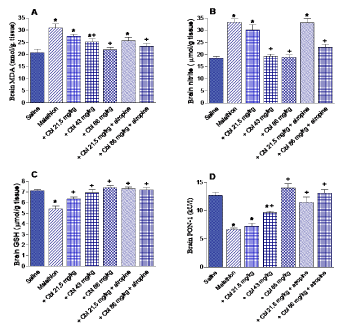
Figure 1. The effect of cerebrolysin (Cbl) given alone or in combination with atropine on the concentration of malondialdehyde (MDA), nitric oxide, reduced glutathione (GSH), and on paraoxonase-1 (PON-1) activity in brain of rats treated with malathion. *: P<0.05 vs corresponding vehicle-treated control group. +: P<0.05 vs corresponding malathion control group.
Nitric oxide
Compared to saline controls, nitric oxide concentration in brain of rats treated with malathion significantly increased by 79.4% (33.24 ± 1.37 vs. 18.54 ± 0.69 mmol/g. tissue). A significant decrease of nitric oxide by 42.2-43.3% was observed after treatment with cerebrolysin at 43 or 86 mg/kg, respectively, as compared to the malathion only group (19.2 ± 0.87 and 18.85 ± 0.96 vs. 33.24 ± 1.37 mmol/g.tissue). Meanwhile, rats given cerebrolysin at 86 mg/kg together with atropine exhibited 31% decrease in brain nitric oxide compared to control value (22.97 ± 1.18 vs. 33.24 ± 1.37 mmol/g. tissue) (Figure 1B).
Reduced glutathione
In malathion only treated rats, reduced glutathione in brain decreased by 24% compared to the saline control values (5.42 ± 0.27 vs. 7.13 ± 0.11 mmol/g. tissue). Reduced glutathione levels were restored by the administration of 86 mg/kg cerebrolysin to the malathion-treated animals (Figure 1C).
Paraoxonase-1 activity
Malathion caused a significant decline in PON-1 activity by 47.6% compared to the saline group (6.65 ± 0.28 vs. 12.7 ± 0.56 kU/l). Cerebrolysin given at 43 or 86 mg/kg resulted in a significant increase in PON-1 activity by 45.1-110.5% compared to the malathion only group (9.65 ± 0.23 and 14.0 ± 0.73 vs. 6.65 ± 0.28 kU/l). The administration of cerebrolysin at 21.5 or 86 mg/kg together with atropine resulted in 71.4-79% increase in brain PON-1 activity compared to malathion control value (11.4 ± 0.91 and 13.1 ± 0.64 vs. 6.65 ± 0.28 kU/l) (Figure 1D).
Acetylcholinesterase activity
Brain AChE activity showed 83.8% decrease in malathion treated rats with respect to control value (1.29 ± 0.08 vs. 7.99 ± 0.41 mmol SH/g/min). A dose-dependent increase in AChE activity (by 172.9, 375.2 and 413.9%) was observed after treatment with cererbrolysin at 21.5, 43 or 86 mg/kg, respectively (3.52 ± 0.22, 6.13 ± 0.18 and 6.63 ± 0.25 vs. 1.29 ± 0.08 mmol SH/g/min). Meanwhile, cerebrolysin given at 21.5 or 86 mg/kg along with atropine resulted in 248.8 and 479.1% increments in AChE activity with respect to control value (4.5 ± 0.18 and 7.47 ± 0.3 vs. 1.29 ± 0.08 mmol SH/g/min) (Figure 2A).
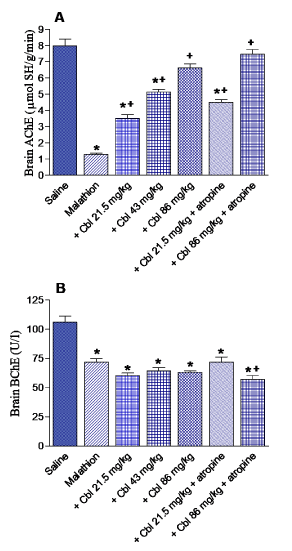
Figure 2. The effect of cerebrolysin (Cbl) given alone or in combination with atropine on the acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) activities in brain of rats treated with malathion. *: P<0.05 vs corresponding vehicle-treated control group. +: P<0.05 vs corresponding malathion control group.
Butryrylcholinesterase activity
A significant decrease in BChE activity by 32.2% was observed in the brain of rats treated with malathion (71.91 ± 3.1 vs. 106 ± 5.7 U/l). There was no significant effect for cerebrolysin given alone on BChE activity. Cerebrolysin given at 86 mg/kg along with atropine, however, caused further decrease in BChE activity by 21% compared with the malathion only group (56.8 ± 3.7 vs. 71.91 ± 3.1 U/l) (Figure 2B).
Histopathological results
Microscopic examination for brain tissue from a control rat showed the normal structure of the cerebral cortex being formed of the innermost granular layer, the central Purkinje cell layer, and the outermost molecular layer (Figure 3A). Rats treated with only malathion showed signs of neurodegeneration (gliosis), thrombotic vessels (vessel with membrane bound vacuoles). Some neurons appeared degenerated; others appeared dark, with hemorrhage in meninges above the surface. Multiple focal homogenous deeply eosinophilic plaques of various sizes and shapes were observed in the cerebral cortex (Figure 3B & 3C). Rats treated with malathion along with cerebrolysin at 21.5 mg/kg showed some improvement in pathological changes in the form of no gliosis, and absent eosinophilic plaques. The granular layer more or less appeared normal. Congestion of many cerebral blood capillaries and dark neurons were seen (Figure 3D). Rats subjected to malathion along with cerebrolysin at 43 mg/mg displayed normal granular cell layer, but congestion of cerebral blood vessel was still present (Figure 3E). After treatment with both malathion and cerebrolysin at 86 mg/kg there was increase in the thickness of the granular layer, and the brain tissue appeared more or less normal (Figure 3F). In rats treated with 24.1 mg/kg malathion along with cerebrolysin and atropine, examination of brain tissue showed increase in the thickness of granular layer and congestion of cerebral blood capillaries (Figure 1G). Sections of brain tissue of rats treated with 86.0 mg/kg cerebrolysin and atropine showed normal architecture (Figure 3F).
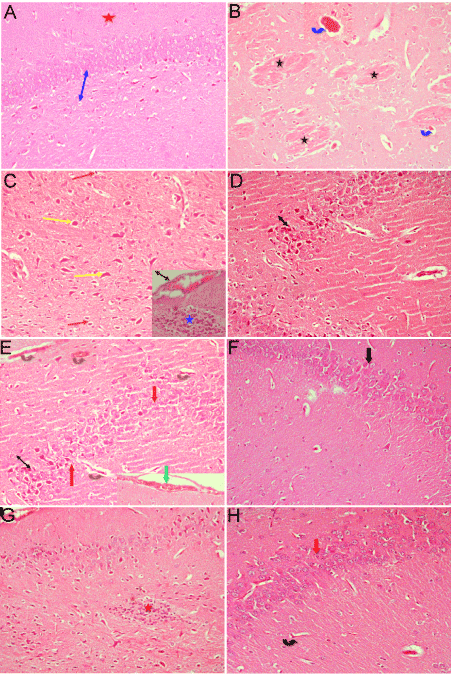
Figure 3. Representative photomicrographs of brain tissue from rats treated with: (A) Saline showing the normal structure. (B) Malathion only showing thrombotic vessels; a vessel with membrane bound vacuoles (blue arrow), and multiple numbers of focal homogenous deeply eosinophilic plaques of various sizes and shapes (star). (C) Malathion only showing neurodegeneration (gliosis) (star). Degenerated (red arrow), and dark neurons (yellow arrow) and hemorrhage in meninges above the surface. (D) Malathion + with cerebrolysin 21.5 mg/kg showing some granular cells being normal but still a decrease in granular layer thickness and degenerated neurons (arrow) (Hx&Ex200).
Representative photomicrographs of brain tissue from rats treated with: (E) Malathion + cerebrolysin 43 mg/kg showing more or less normal granular layer appeared (red arrow) although decreased in thickness, congestion of cerebral blood vessel (brown arrow) and hemorrhage in meninges above the surface (green arrow). (F) Malathion +cerebrolysin 86 mg/kg showing increase in thickness of granular layer (black arrow). (G) Malathion + cerebrolysin 21.5 mg/kg + atropine showing aggregate of gliosis (star) and hemorrhage in meninges above the surface (black arrow). The granular cells appeared normal but still decreased in thickness. (H) Malathion + cerebrolysin 86 mg/kg + atropine showing normal granular cells (red arrow) but congestion of cerebral blood vessels still present (black arrow) (Hx&Ex200).
Discussion
The results of the present study provide the evidence for the first time that treatment with cerebrolysin was able to protect the rat brain against the biochemical changes and neuronal injury caused by acute malathion exposure. In this study and in agreement with previously published data [15,16,42-44], malathion resulted in significant increase in the brain lipid peroxidation end product malondialdehyde concentration, indicative of free radical attack on membrane polyunsaturated fatty acids [20]. There was also a decrease in brain reduced glutathione following malathion intoxication. The tripeptide glutathione (L-g-glutamyl-L-cysteinyl-glycine) functions as a direct scavenger of reactive oxygen metabolites eg., hydroxyl radical and peroxyl radical and reduces hydrogen peroxide and lipid peroxide via glutathione peroxidase-catalyzed reactions [45,46]. In these reactions, the thiol-reduced glutathione form is oxidized to form disulfide-oxidized form (GSSG). The latter is subsequently reduced back to its reduced form by the glutathione reductase using NADPH. The highest concentration of glutathione is found in the mammalian brain tissue being present mainly in its reduced form and hence is considered the most important brain antioxidant [45,47]. The decline in reduced glutathione concentration observed in the brain of rats exposed to malathion is thus most likely to reflect consumption of antioxidant molecule by the generation of free radicals by the toxicant. Other studies indicated increased lipid peroxidation [48,49] and decreased activities of glutathione peroxidae and glutathione reductase [50] in the brain of malathion-treated rats. Increased lipid peroxidation and decreased activities of antioxidant enzymes have also been found in rat or human erythrocytes following malathion exposure in vitro [51,52]. Reactive oxygen metabolites are thus involved in organophosphate neurotoxicty [48-52]. Malathion is capable of increasing the formation of formation of superoxide anion in hippocampal sub-mitochondrial particles. Inhibition of mitochondrial complexs I and IV activities were also observed after malathion application [19,49]. One of the consequences of mitochondrial damage is the increase in the generation of reactive oxygen metabolites eg., the superoxide anion. Reactive oxygen metabolites can then cause mitochondrial damage, thereby, impairing the cell’s bioenergitics [52,53]. Here we show that cerebrolysin was able to decrease brain lipid peroxidation and to increase the level of reduced glutathione, thereby, suggesting that cerebrolysin exerts an antioxidant effect.
As indicated in this study, malathion caused increased brain nitric acid content which is in agreement with previously published data [43,44]. The short-lived gaseous free radical nitric oxide (NO•) is an important signaling molecule and a vasodilator [54]. It is synthesized from L-arginine by the action of the enzyme nitric oxide synthase which exists in an endothelial NO synthase (eNOS), inducible NO synthase (iNOS), and neuronal NO synthase (nNOS) isoforms. While constitutively formed nitric oxide from nNOS and eNOS account for the formation of the low concentrations necessary for normal cellular functions, high concentrations generated for longer time by iNOS are neurotoxic [55]. This is caused by the increased expression of iNOS in microglia cells and astrocytes following their stimulation by inflammatory cytokines and reactive oxygen species [55,56]. Nitric oxide can react with molecular oxygen resulting in the formation of reactive nitrogen oxide species such as dinitrogen trioxide, and nitrogen dioxide and the consequent nitrosylation and/or nitration of proteins and lipids. Nitric oxide also reacts with the superoxide anion, resulting in the formation of the highly reactive peroxynitrite anion (ONOO−) [54,57]. The role of nitric oxide in the development of malathion neurotoxicity is supported by the finding that inhibition of endgenous nitric oxide formation by the non-selective NOS inhibitor NG-nitro-L-arginine methyl ester or the selective neuronal NOS inhibitor 7-nitroindazole was able to reduce brain oxidative stress and neurodegeneration caused by acute malathion in rats [58]. The results of the present study show that treatment with cerebrolysin results in decreased brain nitric oxide oxide which could be involved at least in part in the neuroprotective effects of the drug.
Moreover, and accordance with previous studies [43,44,58], there was markedly decreased PON-1 activity in brain of malathion-intoxicated rats. Meanwhile, treatment with cerebrolysin was shown to increase PON-1 activity. This enzyme is important in the hydrolysis of the active metabolites of a number of organophosphate insecticides eg., paraxon, chlorpyrifos oxon, diazinon oxon as well as nerve agents [59]. Mice deficient in the enzyme displayed increased sensitivity to the toxic actions of chlorpyrifos oxon, and diazoxon [60,61]. In contrast human purified PON-1 injected into PON-1 null mice protected against the decline in AChE activity caused by paraoxon or chlorpyrifos oxon [61]. Furthermore, pretreatment of rats with purified rabbit PON-1 resulted in less inhibition in AChE activity and decreased toxicity caused by dichlorvos [62]. The important role of this enzyme also derives from the evidence that (i) exposure to organophosphate insecticides could be a causative factor in the development of such neurodegenerative diseases eg., Parkinson’s disease in susceptible individuals [10,11] and; (ii) variation in the catalytic efficiency of the enzyme appear to underlie such risk [12].
Organophosphorus compounds irreversibly bind to and inactivate cholinesterases [3,4]. While the role of AChE in neurotransmission and in the toxicity caused by organophosphate insecticides is clear, little is known as regards the role of BChE [4]. There is evidence, however, that BChE acts to protect nerve terminals from increased concentrations of acetylcholine at the neuromuscular junction [63]. Our data demonstrate marked decrease in brain AChE and BChE activities following the administration of malathion. Cerebrolysin given in conjunction with malathion was shown to significant protection against the decline in AChE activity. The drug has also been found to result in less inhibition in AChE activity in the cerebral cortex in rats treated with rotenone [35]. Whether this effect of cerebrolysin is a consequence of a neuroprotective action or due to reactivation of the enzyme is yet to be determined.
The neuroprotective action of cerebrolysin against acute malathion toxicity was also confirmed by histopathological studies of the brain tissue which revealed gliosis, degenerated and dark neurons and eosinophilic plaques in the cerebral cortex of the malathion control group but not in the cerebrolysin-treated groups. Cerebrolysin also increased the thickness of the granular layer denoting. This histological improvement was also observed when cerebrolysin and atropine were combined, suggesting that both drugs could be used safely in the treatment of acute malathion toxicity in humans.
In summary, the present study indicated significantly increased brain oxidative stress and the development of neurodegeneration following acute malathion exposure. These effects of the neurotoxicant were ameliorated by cerebrolysin given alone or in conjunction with atropine with restoration of AChE activity. Cerebrolysin might thus prove useful in the management of neurotoxicity due to malathion and other organophosphates.
Acknowledgements
This work is not supported by research grants
Conflicts of interest
The authors declare that there are no conflicts of interest
References
- Jokanovic M, Kosanovic M (2010) Neurotoxic effects in patients poisoned with organophosphorus pesticides. Environ Toxicol Pharmacol 29: 195-201. [crossref]
- Maroni M, Colosio C, Ferioli A, Fait A (2000) Biological monitoring of pesticide exposure: a review–. introduction. Toxicology 143: 1–118. [crossref]
- Mileson BE, Chamber2021 Copyright OAT. All rights reservet al. (1998) Common mechanism of toxicity: a case study of organophosphorus pesticides. Toxicol Sci 41: 8-20. [crossref]
- Mileson BE, Chambers JE, Chen WL, Dettbarn W, Ehrich M, et al. (1998) Common mechanism of toxicity: a case study of organophosphorus pesticides. Toxicol Sci 41: 8-20. [crossref]
- Silman I, Sussman JL (2005) Acetylcholinesterase: 'classical' and 'non-classical' functions and pharmacology. Curr Opin Pharmacol 5: 293-302. [crossref]
- Blain PG (2011) Organophosphorus poisoning (acute). BMJ Clin Evid 2011. [crossref]
- Michotte A, Van Dijck I, Maes V, D’Haenen H (1989) Ataxia as the only delayed neurotoxic manifestation of organophosphate insecticide poisoning. Eur Neurol 29: 23–26. [crossref]
- Lotti M, Moretto A (2005) Organophosphate-induced delayed polyneuropathy. Toxicol Rev 24: 37-49. [crossref]
- Harrison V, Ross SM (2016) Anxiety and depression following cumulative low-level exposure to organophosphate pesticides. Environ Res 151: 528–536. [crossref]
- Jokanovic M, Stukalov PV, Kosanovic M (2002) Organophosphate induced delayed polyneuropathy. Curr Drug Targets CNS Neurol Disord 1: 593-602. [crossref]
- Hancock DB, Martin ER, Mayhew GM, Stajich JM, Jewett R, et al. (2008) Pesticide exposure and risk of Parkinson's disease: a family-based case-control study. BMC Neurol 8: 6. [crossref]
- Pezzoli G, Cereda E (2013) Exposure to pesticides or solvents and risk of Parkinson disease. Neurology 80: 2035-2041. [crossref]
- Paul KC, Sinsheimer JS, Cockburn M, Bronstein JM, Bordelon Y, et al. (2017) Organophosphate pesticides and PON1 L55M in Parkinson's disease progression. Environ Int 107: 75-81. [crossref]
- Manthripragada AD, Costello S, Cockburn MG, Bronstein JM, Ritz B (2010) Paraoxonase 1, agricultural organophosphate exposure, and Parkinson disease. Epidemiology 21: 87-94. [crossref]
- Lee PC, Rhodes SL, Sinsheimer JS, Bronstein J, Ritz B (2013) Functional paraoxonase 1 variants modify the risk of Parkinson's disease due to organophosphate exposure. Environ Int 56: 42-47. [crossref]
- Moore PD, Yedjou CG, Tchounwou PB (2010) Malathion-induced oxidative stress, cytotoxicity, and genotoxicity in human liver carcinoma (HepG2) cells. Environ Toxicol 25: 221-226. [crossref]
- Edwards FL, Yedjou CG, Tchounwou PB (2013) Involvement of oxidative stress in methyl parathion and parathion-induced toxicity and genotoxicity to human liver carcinoma (HepG2) cells. Environ Toxicol 28: 342-348. [crossref]
- Kaur P, Radotra B, Minz RW, Gill KD (2007) Impaired mitochondrial energy metabolism and neuronal apoptotic cell death after chronic dichlorvos (OP) exposure in rat brain. Neurotoxicology 28: 1208-1219. [crossref]
- Middlemore-Risher ML, Adam BL, Lambert NA, Terry AV Jr (2011) Effects of chlorpyrifos and chlorpyrifos-oxon on the dynamics and movement of mitochondria in rat cortical neurons. J Pharmacol Exp Ther 339: 341-349. [crossref]
- Karami-Mohajeri S, Hadian MR, Fouladdel S, Azizi E, Ghahramani MH, et al. (2014) Mechanisms of muscular electrophysiological and mitochondrial dysfunction following exposure to malathion, an organophosphorus pesticide. Hum Exp Toxicol 33: 251-263. [crossref]
- Halliwell B (2007) Biochemistry of oxidative stress. Biochem Soc Trans 35: 1147-1150. [crossref]
- Sies H (1997) Oxidative stress: oxidants and antioxidants. Exp Physiol 82: 291-295. [crossref]
- Halliwell B (2001) Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging 18: 685-716. [crossref]
- Roberts DM, Aaron CK (2007) Management of acute organophosphorus pesticide poisoning. BMJ 334: 629-634. [crossref]
- Eddleston M, Szinicz L, Eyer P, Buckley N (2002) Oximes in acute organophosphorus pesticide poisoning: a systematic review of clinical trials. QJM 95: 275-283. [crossref]
- Buckley NA, Eddleston M, Li Y, Bevan M, Robertson J (2011) Oximes for acute organophosphate pesticide poisoning. Cochrane Database Syst Rev 2: CD005085. [crossref]
- Masliah E, Díez-Tejedor E (2012) The pharmacology of neurotrophic treatment with Cerebrolysin: brain protection and repair to counteract pathologies of acute and chronic neurological disorders. Drugs Today (Barc) 48: 3-24. [crossref]
- Allegri RF, Guekht A (2012) Cerebrolysin improves symptoms and delays progression in patients with Alzheimer's disease and vascular dementia. Drugs Today (Barc) 48: 25-41. [crossref]
- Guekht AB, Moessler H, Novak PH, Gusev EI (2011) Cerebrolysin Investigators. Cerebrolysin in vascular dementia: improvement of clinical outcome in a randomized, double-blind, placebo-controlled multicenter trial. J Stroke Cerebrovasc Dis 20: 310-318. [crossref]
- Amiri-Nikpour MR, Nazarbaghi S, Ahmadi-Salmasi B, Mokari T, Tahamtan U, et al. (2014) Cerebrolysin effects on neurological outcomes and cerebral blood flow in acute ischemic stroke. Neuropsychiatr Dis Treat 10: 2299–2306. [crossref]
- Hartbauer M, Hutter-Paie B, Windisch M (2001) Effects of Cerebrolysin on the outgrowth and protection of processes of cultured brain neurons. J Neural Transm (Vienna) 108: 581-592. [crossref]
- Zhang C, Chopp M, Cui Y, Wang L, Zhang R, et al. (2010) Cerebrolysin enhances neurogenesis in the ischemic brain and improves functional outcome after stroke. J Neurosci Res 88: 3275-3281. [crossref]
- Hartbauer M, Hutter-Paier B, Skofitsch G, Windisch M (2001) Antiapoptotic effects of the peptidergic drug cerebrolysin on primary cultures of embryonic chick cortical neurons. J Neural Transm (Vienna) 108: 459-473. [crossref]
- Juárez I, González DJ, Mena R, Flores G (2011) The chronic administration of cerebrolysin induces plastic changes in the prefrontal cortex and dentate gyrus in aged mice. Synapse 65: 1128-1135. [crossref]
- Abdel-Salam OM, Omara EA, Mohammed NA, Youness ER, Khadrawy YA, et al. (2013) Cerebrolysin attenuates cerebral and hepatic injury due to lipopolysaccharide in rats. Drug Discov Ther 7: 261-271. [crossref]
- Abdel-Salam OME, Mohammed NA, Youness ER, Khadrawy YA, Omara EA, et al. (2014) Cerebrolysin protects against rotenone-induced oxidative stress and neurodegeneration. J Neurorestoratol 2: 47–63.
- Nair V, Turner GA (1984) The thiobarbituric acid test for lipid peroxidation: structure of the adduct with malondialdehyde. Lipids 19: 804-805.
- Archer S (1993) Measurement of nitric oxide in biological models. FASEB J 7: 340-360. [crossref]
- Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82: 70-77. [crossref]
- Eckerson HW, Wyte CM, La Du BN (1983) The human serum paraoxonase/arylesterase polymorphism. Am J Hum Genet 35: 1126-1138. [crossref]
- Haagen L, Brock A (1992) A new automated method for phenotyping arylesterase (EC 3.1.1.2) based upon inhibition of enzymatic hydrolysis of 4-nitrophenyl acetate by phenyl acetate. Eur J Clin Chem Clin Biochem 30: 391-395. [crossref]
- Ellman GL, Courtney KD, Andres V Jr, Feather-Stone RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7: 88-95. [crossref]
- Gorun V, Proinov I, Baltescu V, Balaban G, Barzu O (1978) Modified Ellman procedure for assay of cholinesterases in crude enzymatic preparations. Anal Biochem 86: 324-326. [crossref]
- Abdel-Salam OME, Youness ER, Esmail RSE, Mohammed NA, Khadrawy YA, et al. (2016) Methylene blue as a novel neuroprotectant in acute malathion intoxication. Reactive Oxygen Species 1: 165–177.
- Abdel-Salam OM, Youness ER, Mohammed NA, Yassen NN, Khadrawy YA, et al. (2016) Novel neuroprotective and hepatoprotective effects of citric acid in acute malathion intoxication. Asian Pac J Trop Med 9: 1181-1194. [crossref]
- Mytilineou C, Kramer BC, Yabut JA (2002) Glutathione depletion and oxidative stress. Parkinsonism Relat Disord 8: 385–387. [crossref]
- Lu SC (2013) Glutathione synthesis. Biochim Biophys Acta 1830: 3143-3153. [crossref]
- Wu G, Fang YZ, Yang S, Lupton JR, Turner ND (2004) Glutathione metabolism and its implications for health. J Nutr 134: 489-492. [crossref]
- Brocardo PS, Pandolfo P, Takahashi RN, Rodrigues AL, Dafre AL (2005) Antioxidant defenses and lipid peroxidation in the cerebral cortex and hippocampus following acute exposure to malathion and/or zinc chloride. Toxicology 207: 283-291. [crossref]
- Delgado EH, Streck EL, Quevedo JL, Dal-Pizzol F (2006) Mitochondrial respiratory dysfunction and oxidative stress after chronic malathion exposure. Neurochem Res 31: 1021–1025. [crossref]
- Trevisan R, Uliano-Silva M, Pandolfo P, Franco JL, Brocardo PS, et al. (2008) Antioxidant and acetylcholinesterase response to repeated malathion exposure in rat cerebral cortex and hippocampus. Basic Clin Pharmacol Toxicol 102: 365-369. [crossref]
- John S, Kale M, Rathore N, Bhatnagar D (2001) Protective effect of vitamin E in dimethoate and malathion induced oxidative stress in rat erythrocytes. J Nutr Biochem 12: 500-504. [crossref]
- Durak D, Uzun FG, Kalender S, Ogutcu A, Uzunhisarcikli M, et al. (2009) Malathion-induced oxidative stress in human erythrocytes and the protective effect of vitamins C and E in vitro. Environ Toxicol 24: 235-242. [crossref]
- Lenaz G (2001) The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life 52: 159-164. [crossref]
- Genova ML, Pich MM, Bernacchia A, Bianchi C, Biondi A, et al. (2004) The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann N Y Acad Sci 1011: 86-100. [crossref]
- Pacher P, Beckman JS, Liaudet L (2007) Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87: 315-424. [crossref]
- Guix FX, Uribesalgo I, Coma M, Muñoz FJ (2005) The physiology and pathophysiology of nitric oxide in the brain. Prog Neurobiol 76: 126-152. [crossref]
- Förstermann U, Sessa WC (2012) Nitric oxide synthases: regulation and function. Eur Heart J 33: 829-837. [crossref]
- Thomas DD, Miranda KM, Citrin D, Espey MC, Wink DA (2003) Nitric Oxide. In: Combat Medicine: Basic and Clinical Research in Military, Trauma, and Emergency Medicine. Tsokos Gc, Atkins JL (Eds) Humana Press Inc., Totowa, NJ, 23-60.
- Abdel-Salam OME, Youness ER, Mohammed NA, Yassen NN, Khadrawy YA, et al. (2017) Nitric oxide synthase inhibitors protect against brain and liver damage caused by acute malathion intoxication. Asian Pac J Trop Med 10: 773–786. [crossref]
- La Du BN (1992) Human serum paraoxonase/arylesterase. In Pharmacogenetics of Drug Metabolism. Kalow W (Ed.). Pergamon Press, Inc., New York, 51-91.
- Costa LG, Li WF, Richter RJ, Shih DM, Lusis A, et al. (1999) The role of paraoxonase (PON1) in the detoxication of organophosphates and its human polymorphism. Chem Biol Interact 119: 429-438. [crossref]
- Li WF, Costa LG, Richter RJ, Hagen T, Shih DM, et al. (2000) Catalytic efficiency determines the in-vivo efficacy of PON1 for detoxifying organophosphorus compounds. Pharmacogenetics 10: 767-779. [crossref]
- Wang NN, Dai H, Yuan L, Han ZK, Sun J, et al. (2010) Study of paraoxonase-1 function on tissue damage of dichlorvos. Toxicol Lett 196: 125-132. [crossref]
- Girard E, Bernard V, Minic J, Chatonnet A, Krejci E, et al. (2007) Butyrylcholinesterase and the control of synaptic responses in acetylcholinesterase knockout mice. Life Sci 80: 2380-2385. [crossref]