An adult male patient came to our observation for a brain magnetic resonance imaging (MRI) study because of progressive symptoms characterized by ataxia, tremor, rigidity and cognitive dysfunction. The neuropsychological tests confirmed an IQ reduction and cognitive impairment. The MRI study showed symmetric T1 decreased and T2 increased signal intensity in cerebellar white matter (wm) lateral, superior, and inferior to the cerebellar dentate nuclei. Moreover, hyperintense T2 focal areas of both deep wm cerebral hemisphere and T2 hyperintensity of the splenium of the corpus callosum were documented. The progressive neurological symptoms of our adult male patient and the MRI study features suggested a degenerative disease, due to fragile X premutation. Genetic tests confirmed the diagnostic hypothesis of fragile X-associated tremor ataxia syndrome (FXTAS).
movement disorder, fragile X tremor Ataxia Syndrome, MRI middle cerebellar peduncle signs
Fragile X syndrome is the most common inherited form of mental retardation, with the carrier frequency in the general population being approximately one in 250 female and one in 760 male persons [1,2]. The disorder is caused by an expansion in excess of 200 repeats (full mutation expansion) of a trinucleotide element, (CGG), located in the 5’ untranslated region of the fragile X mental retardation 1 (FMR1) gene [3]. Full mutation expansions are generally accompanied by silencing of the FMR1 gene, with attendant lack of FMR1 protein (FMRP) synthesis; it is the lack of FMRP that leads to fragile X syndrome. Trinucleotide expansions that are in the 55to 200 repeat range are termed permutations. Healthy subjects may have a range of approximately five to 54 repeats. Carriers of the FMR1premutation allele (55-199 repeats as defined by the American College of Medical Genetics [4] are at significant risk for a late-onset neurodegenerative disorder, fragile X-associated tremor ataxia syndrome (FXTAS) [5]. This disorder is distinct from that manifested in carriers of the full mutation (> 200 repeats), or fragile X syndrome (FXS), with respect to the molecular etiology and clinical phenotype. The primary features of FXTAS are action tremor and gait ataxia. Associated features include parkinsonism, autonomic dysfunction, peripheral neuropathy, and cognitive dysfunction. Cognitive changes are highly variable and include global impairments as well as specific executive cognitive dysfunction [6-7]. Preliminary reports suggest that psychiatric symptoms include anxiety, agitation, disinhibition, and depression. This characteristic pattern of cognitive and behavioral deficits resembles that observed in other forms of frontal–subcortical dementia [8]. Neuro-imaging features of FXTAS on T2 weighted brain MRI include middle cerebellar peduncles (MCP) and periventricular, subcortical white matter (wm) changes [9]. The MCP hyperintensity has been incorporated in the diagnostic criteria for “definite” FXTAS [10]. Though distinctive, it is found in only about 64% of individuals with FXTAS [11]. Other important radiological findings are cerebral and cerebellar cortical atrophy, thinned corpus callosum, reduction of both middle cerebellar peduncles sagittal diameter and pontine transverse/rostral-caudal dimension [12]. These radiological findings in an elderly patient with ataxia, tremor, rigidity and cognitive dysfunction, in addition to mandatory genetic specific tests, may improve the FXTAS diagnosis.
A 63 years old Caucasian male affected by progressive and worsening intention tremor, gait ataxia, autonomic dysfunction, rigidity and cognitive decline came to our observation. Neuropsychological tests confirmed the reduction of IQ and impaired executive functions. The MRI brain study (Figures 1 and 2) showed pathologic symmetric signal of wm of the middle cerebellar peduncles (MCPs), specifically localized medially, superiorly and laterally to the dentate nuclei, showing restricted Diffusion on specific sequences. The MCPs transverse diameter was reduced. The infra-tentorial subarachnoid spaces were enlarged due to cerebellar cortical atrophy. The pons and the midbrain also appeared atrophic, both in axial and sagittal planes, with enlargement of the interpeduncular cistern and concave appearance of the midbrain, but Parkinsonism Index was not diagnostic for Progressive Supranuclear Palsy (PSP) or Multiple System Atrophy (MSA). The MRI documented cortico-subcortical brain atrophy, especially in frontoparietal regions and hyperintense T2 focal areas of both deep wm cerebral hemisphere and T2 hyperintensity of the splenium of the corpus callosum. The corpus callosum also appeared with a reduced thickness. The Evans index was normal, but the third ventricle was dilated. Both MRI findings and the worsening of neurologic symptoms suggested an adult-fragile X premutation. The genetic tests, conducted with Southern blot and PCR, showed an expansion of CGG triplets in the 5' untranslated region of the Fragile X Mental Retardation gene. At the end, a defined diagnosis of the fragile X-associated tremor ataxia syndrome (FXTAS) carrier was made.
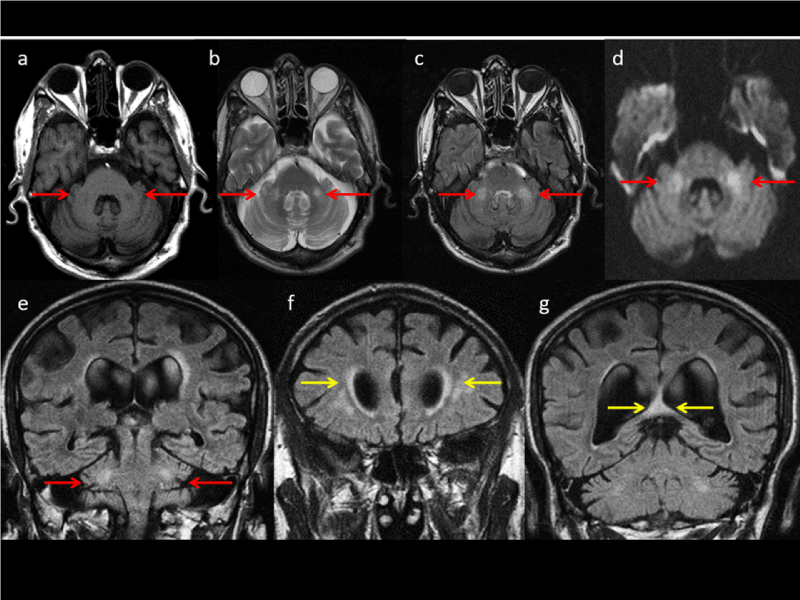
Figure 1. Figure 1. Axial (a, b, c and d) and Coronal MRI study shows pathologic signal of middle cerebellar peduncles; note hypointense onT1 (a) and hyperintense signal on T2 (b, c and e) and Diffusion sequences (d) (see red head arrows). Small and confluent deep white matter T2 hyperintense area respectively in frontal region (f) and hyperintense focal T2 hyperintense area of the splenium of corpus callosum on FLAIR weighted sequence (g) (see yellow head arrows)
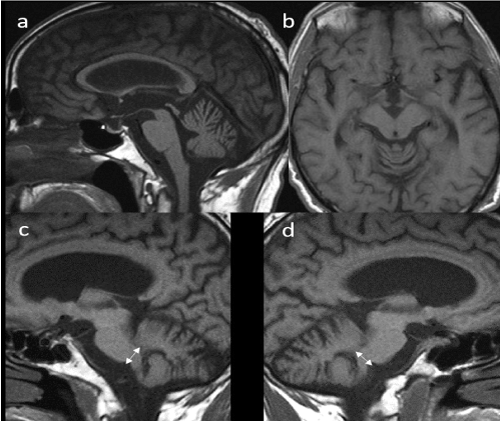
Figure 2. Sagittal (a, c and d) and transverse (b) MRI T1 sequences. Diffuse enlargement of peri cerebellar sulci, mild atrophy of the pons, corpus callosum and mesencephalon (a) with slight enlargement of interpeduncular cistern (b) and reduction in diameter of the middle cerebellar peduncles (c and d)
Fragile X syndrome is caused by absence or deficiency of the Fragile X mental retardation protein (FMRP), a protein that is crucial for early brain development, maturation and plasticity [1-2]. Although the focus of fragile X syndrome has been on its relationship to early child development, a newly recognized disorder, associated with fragile X premutation, has been noted to manifest in elderly men as the presence of tremor, ataxia, dyssynergia, cognitive decline, rigidity, and impotence. The FXTAS represents this disorder [5]. The diagnostic criteria of FXTAS comprise three domains: clinical, radiological, and pathological [13]. Whether definite, probable or possible, FXTAS diagnosis requires a degree of functional/clinical impairment. While the clinical presentation is variable, the initial presenting motor symptom is often an intention or kinetic tremor, followed by cerebellar ataxia, both of which typically progress over time [14]. Other clinical features may include executive dysfunction, ranging from mild to dementia-like in severity, which often presents later in the disease course [15]. The most prominent neuropathological features of FXTAS are: (1) cerebral and cerebellar wm disease that is pathologically distinct from that found in cerebrovascular and demyelinating disorders, (2) astrocytic pathology in wm, and (3) eosinophilic, ubiquitin-positive intranuclear inclusions in brain and spinal cord [16,17].Currently, FXTAS neuropathology can only be confirmed after death and hence cannot be used as a prognostic tool. The radiological phenotype emphasizes FXTAS as a wm neurodegenerative disease, as prior studies have highlighted the presence of wm lesions in the MCPs, known as the MCPs sign, and brainstem, as well as generalized cerebral lesions and atrophy throughout the neocortex [18-20]. Indeed, it has been suggested that hyperintensity of the splenium of the corpus callosum should be added as a minor criterion for FXTAS, given that these lesions were found to be more common than MCPs lesions in female premutation carriers with FXTAS [21]. The MCPs sign is not specific to FXTAS [22] and is occasionally seen in other rare neurodegenerative diseases. With the exception of the required premutation allele, none of the clinical features are absolutely specific for FXTAS [23].Although the MR findings of symmetrically increased T2 signal intensity within the MCPs and adjacent cerebellar wm have been shown in this study to be characteristic of symptomatic elderly men with fragile X premutation, these findings are not specific. The phenotype of FXTAS is very similar to that of other neurodegenerative disorders (ND). The movement abnormalities seen in FXTAS overlap with those of several more well-known disorders. In 2016, Robertson, et al. published a systematic review in which they compared the phenotype of FXTAS to that of essential tremor (ET), Parkinson’s disease (PD), spinocerebellar ataxias (SCAs), multiple system atrophy (MSA) and progressive supranuclear palsy (PSP) [23]. Although patients with FXTAS show complex clinical phenotypes, features such as parkinsonism can make it difficult to distinguish from persons with Parkinson’s disease [24]. Symptoms early in the disease course may be mild and difficult to categorize. Individuals with minor unsteadiness and tremor may be diagnosed with essential tremor. It is important to be aware that patients with essential tremor who develop unsteadiness and/or executive dysfunction, may have alternative diagnoses, including FXTAS. This also applies to cases with atypical development of other movement disorders. Although clinical manifestations are essential for placing a correct diagnosis, morphological MRI imaging plays a fundamental role in the diagnosis and follow-up of patients with ND. However, since different diseases of this heterogeneous group may have overlapping morphological elements, the interpretation of the image must be performed only in correlation with the clinical presentation of the patient [25]. In FXTAS, action tremor can be present alone or in association with resting tremor. Half of all patients can show cerebellar ataxia, while parkinsonism can be present in around one third of them. We can also find other symptoms like cognitive impairment with executive dysfunction, neuropathy and sleep disturbances [26]. In ET symptoms like cerebellar ataxia and parkinsonism have not been reported (only minor cerebellar abnormalities later in the disease course have been described) and the MCPs sign is not present [26]. PD is characterized by asymmetrical resting tremor, most often in hands, rigidity, bradykinesia, postural instability and no motor symptoms like sleep disorders, depression and reduced olfactory sense [26]. Patients with symptoms of idiopathic primary PD have not specific diagnostic neuroradiological findings, therefore the diagnosis is exclusively based on the clinical signs and the purpose of the MRI study consists in the overall assessment of regressive-atrophic encephalic phenomena, of the extent of any signs of subcortical ischemic encephalopathy and finally in the exclusion of secondary forms [27]. Movement disorder in SCAs is characterized by intention tremor and slowly progressive ataxia. Unsteady gait is the first symptom. Parkinsonism may be seen in some SCA type. Examination of eye movements often reveals fragmented, jerky trekking movements and slow saccades [26]. SCA VI with clinical symptoms of an autosomal dominant cerebellar ataxia type III has been described as occurring in a single patient in association with increased T2 signal intensity in the MCPs and mild atrophy of the pons and cerebellum [28]. Three separately reported cases of SCA VI have not had the alteration in posterior fossa white matter T2 signal intensity [29]. Patients with SCA type I, II, III, V, VIII, XII, and XIII have been shown to have volume loss of varying severity involving the cerebellar hemispheres, vermis, and pons [30-36]. In PSP tremor and cerebellar ataxia are not usually present, while we can find parkinsonism characterized by axial rigidity, vertical gaze palsy, slow saccades and executive dysfunction [26]. The MRI finding is characterized by the volumetric reduction of the midbrain, predominantly at the tegument, with consequent deepening of the posterior portion of the III ventricle that dilates as a "funnel", like observable in the sagittal scans with thin thickness. When the atrophy is very pronounced the MRI investigation in the axial scans documents the atrophy of the superior colliculi and a deepening of the interpeduncular cistern. With the long TR sequences, it is possible to highlight the astrogliosis as hyperintensities of the periaqueductal region, of the red nuclei and of the Globus pallidus. Because of the increased deposition of iron, the putamina may appear, contrary to the norm, hypointense with respect to the Globus pallidus, in the T2 and T2* weighted sequences [27]. In patients with PSP increased signal intensity has been described as occurring in the superior cerebellar peduncles on proton diffusion-weighted images [37], although normal signal intensity can be seen in the deep cerebellar wm and MCPs. Alterations involving the superior cerebellar peduncles were not shown to occur in the patients with FXTAS. MSA represents three diseases, previously considered as distinct syndrome: Nigro-striatal degeneration (NSD), olivo-Ponto-cerebellar atrophy (OPCA) and Shy-Drager syndrome (SDS). We can speak about forms or variants of MSA: in cases with prevalence of parkinsonian signs the disease is the MSA-p (MSA-Parkinson type, identifiable with DNS), while in cases with prevalent cerebellar and brain-encephalic dysfunction it is the MSA-c (MSA-cerebellar type, identifiable with the OPCA). The SDS, characterized by Parkinsonian type movement disorders and severe dysregulation of the autonomic nervous system, has no reason to exist as a disease, as its characteristics are present in varying degrees in all forms of MSA [27]. The patients with MSA can show tremor, cerebellar ataxia (MSA-c), parkinsonism (MSA-p), other non-motor symptoms like autonomic dysfunction, dystonia and REM sleep disturbance [26]. The findings of the MR study do not have high sensitivity, especially at an early stage, but, when present, they reflect neuropathological alterations and can make an essential contribution to the diagnosis of the specific form of the disease. In MSA-P atrophy, gliotic degeneration and ferritin iron deposition in putamina involves a typical and specific alteration, the linear signal rim (hyperintense in T2 / DP and FLAIR weighted sequences and hypointense in T2* weighted sequences), in correspondence of the postero-lateral portion of the putamen [38]. A careful examination can also reveal a reduction, sometimes asymmetrical, of the transverse diameter of the putamen, and the atrophy of the head of the caudate nucleus, better visible in the coronal plane, with loss of the typical indentation on the lateral wall of the ventricular frontal horns [27].While the MRI findings at the basal ganglia are more frequent in case of MSA-P, or MSA-C (predominantly cerebellar symptomatology), the essential neuropathological element is represented by the degeneration of the pontine nuclei and therefore of the transverse fibers of the pons, cerebellar peduncles, wm of the cerebellar hemispheres and olives. On these cases, sometimes associated with basal ganglia findings already described, a typical picture is observed, characterized by the reduction of the pontine volume and T2 hyperintense signal, which highlights the cross-sectional fibers of the pons which can extend to the average cerebellar peduncles, with a cross arrangement, from which the sign of the “ hot cross buns” [27]. Among the mentioned and described neurodegenerative diseases, MSA type C can mimic clinically FXTAS more than the others one. In fact, Maureen A. Leehey, et al. [39] recommended genetic testing for FMR1 premutation even in suspected cases of MSA type C. In any case, the presence of the MCPs sign can help a correct diagnostic classification in a patient with movement disorders similar to that described in our case. However, on the other hand there are many other diseases with involvement of MCPs: this is the reason why a differential diagnosis needs to be considered adequately. When approaching abnormal T2 signal in the MCP, entities such Multiple Sclerosis (MS), PML (Progressive Multifocal Leukoencephalopathy), PRES (Posterior Reversible Encephalopathy Syndrome) and certain toxic/metabolic or vascular states should be entertained. Careful evaluation of concomitant correlation with key clinical findings discussed would yield an appropriate and accurate differential diagnosis in most cases. Cerebellar symptoms and signs are reported in 50%80% of MS patients and on conventional MRI frequency of brainstem lesions and cerebellar lesions is 68% and 49%88% respectively [40]. Posterior fossa involvement of JC virus has been reported in 58% of cases [41], when present there is MCP involvement in 64%100% of cases [42]. Involvement of the brainstem by PRES has been documented in 18% of cases [43]. Isolated involvement of the brainstem or basal ganglia was seen in 4% of cases (central variant of PRES) [44] and this “atypical distribution patterns” of PRES can be more common than expected [45]. Toxic/drug induced involvement of the MCP has been reported after heroin inhalation (“chasing the dragon”) [46]. Other toxic drug induced abnormalities, such as toluene and methotrexate toxicity, involving the cerebellum and MCP have also been described [47-49]. MCP is supplied by the anterior inferior cerebellar artery (AICA) and in lesser degree by the superior cerebellar artery. In AICA infarction, that represents an uncommon event (thromboembolic or secondary to severe atherosclerotic disease of basilar artery and branches), specific restricted diffusion is seen in the involved cerebellar peduncle. Wallerian Degeneration (WD) in the MCP can occur in the setting of pontine ischemia or hemorrhage. Increased T2 signal caused by gliosis would correspond to the third of four stages in the evolution of WD, as described by Kuhn, et al. [50]. Increased signal on DWI in the corticospinal tract (CST) have been documented in about 20% of cases of MCA/ACA infarcts as early as 72 h [51] and similarly, restricted diffusion in the CST has been reported at 48 h after MCA ischemia in the pediatric population [52] and in the bilateral MCP at 34 week after acute pontine insult [53-56]. In all cases the changes are attributed to early WD, representing different phases in the development of WD and should not be misinterpreted as ischemic insults. At the end the MCPs can be affected in inherited metabolic disorders represented by Leuco Distrophy (LD). However, there is concomitant, usually symmetric involvement of the supratentorial wm, with sparing of subcortical u-fibers [57].
FXTAS is a relatively rare movement disorder with diverse clinical symptoms including motor, sensory, and cognitive findings that first must be clinically valued. The disorder may mimic other ND, especially MSA C. MRI typically reveals generalized brain atrophy, characteristic bilateral MCPs T2 hyper-intensity (often referred to as the MCPs sign), and sometime T2 hyper-intensity of posterior region of the corpus callosum. However, in order to establish a possible, probable and definitive diagnosis of the disease, a genetic test is always necessary to evaluate the presence of the pre-mutation. Finally, it is mandatory a genetic counselling for the patient and her/his family, due to the potential incidence of this genetic syndrome in children born from the female relatives of the patient carrier.
- Rousseau F, Rouillard P, Morel ML, Khandjian EW, Morgan K (1995) Prevalence of carriers of premutation-size alleles of the FMRI gene and implications for the population genetics of the fragile X syndrome. Am J Hum Genet 57: 1006-1018.
- Rousseau F, Morel ML, Rouillard P, Khandjian EW, Morgan K (1997) Surprisingly low prevalence of FMR1 premutation among males from the general population. Am J Hum Genet 59: 188.
- Hagerman RJ, Hagerman PJ (2001) Fragile X syndrome: a model of gene-brain-behaviour relationships. Revista de Neurologia 33: 51-57.
- Sherman S, Pletcher BA, Driscoll DA (2005) Fragile X syndrome: diagnostic and carrier testing. Genet Med 7: 584-587. [Crossref]
- Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, et al. (2001) Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 57: 127-130. [Crossref]
- Grigsby J, Brega AG, Jacquemont S, Loesch DZ, Leehey MA, et al. (2006) Impairment in the cognitive functioning of men with fragile X-associated tremor/ataxia syndrome (FXTAS). J Neurol Sci 248: 227-233.
- Brega AG, Goodrich G, Bennett RE, Hessl D, Engle K, et al. (2008) The primary cognitive deficit among males with fragile X-associated tremor/ataxia syndrome (FXTAS) is a dysexecutive syndrome. J Clin Exp Neuropsychol 30: 853-869.
- Bacalman S, Farzin F, Bourgeois JA, Cogswell J, Goodlin BL, et al. (2006) Psychiatric phenotype of the fragile X-associated tremor/ataxia syndrome (FXTAS) in males: newly described frontosubcortical dementia. J Clin Psychiatry 67: 87-94.
- Brunberg JA, Jacquemont S, Hagerman RJ, Kravis EM, Grigsby J, et al. (2004) Fragile X premutation carriers: characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction. Am J Neuroradiol 23: 1757-1766.
- Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, et al. Fragile X permutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet 72: 869-878.
- Hagerman PJ, Hagerman RJ (2007) Fragile X-associated tremor/ataxia syndrome-an older face of the fragile X gene. Nat Clin Pract Neurol 3:107-112. [Crossref]
- James A, Brunberg S (2002) Fragile X Premutation Carriers: Characteristic MR Imaging Findings of Adult Male Patients with Progressive Cerebellar and Cognitive Dysfunction. AJNR 23: 1757-1766.
- Jacquemont S, Hagerman R, Leehey M, Grigsby J (2003) Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet 72: 869-878.
- Apartis E, Blancher A, Meissner WG (2012) FXTAS: new insights and the need for revised diagnostic criteria. Neurology 79: 1898-1907.
- Seritan AL, Nguyen DV, Farias ST, Hinton L, Grigsby J, et al. (2008) Dementia in fragile X-associated tremor/ataxia syndrome (FXTAS): comparison with Alzheimer’s disease. Am J Med Genet 147: 1138-1144.
- Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, et al. (2006) Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain 129: 243-255.
- Greco CM, Hagerman RJ, Tassone F, Chudley AE, Bigio MR, et al. (2002) Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 125: 1760-1771.
- Cohen S, Masyn K, Adams J, Hessl D, Rivera S, et al. (2006) Molecular and imaging correlates of the fragile X-associated tremor ataxia syndrome. Neurology 67: 1426-1431.
- Rivera S, Stebbins GT, Grigsby J (2010) Radiological Findings in FXTAS, in The Fragile X-Associated Tremor Ataxia Syndrome (FXTAS). Tassone F, Berry-Kravis EM (Eds), Springer Science pp. 55-66.
- Hashimoto R, Javan AK, Tassone F, Hagerman RJ, Rivera SM (2011) A voxel-based morphometry study of grey matter loss in fragile X-associated tremor/ataxia syndrome. Brain 134: 863-878.
- Apartis E, Blancher A, Meissner WG, Guyan L, Maltête D, et al. (2012) FXTAS: new insights and the need for revised diagnostic criteria. Neurology 79: 1898-1907.
- Storey E, Billimoria P (2005) Increased T2 signal in the middle cerebellar peduncles on MRI is not specific for fragile X premutation syndrome. J Clin Neurosci 12: 42-43.
- Robertson EE, Hall DA, McAsey AR (2016) Fragile X-associated tremor/ataxia syndrome: phenotypic comparisons with other movement disorders. Clin Neuropsychol 30: 849-900.
- Hall DA, Howard K, Hagerman R (2009) Parkinsonism in FMR1 premutation carriers may be indistinguishable from Parkinson disease. Parkinsonism Relat Disord 15: 156-159. [Crossref]
- Conforti R, Capasso R, Negro A (2013) Diagnostic tools in neurodegenerative disorders of adult-elderly. Recenti Prog Med 104: 295-298.
- Hoem G, Koht J (2017) Fragile X-associated tremor/ataxia syndrome. Tidsskr Nor Laegeforen 137.
- Colosimo C, Riario S (2003) La diagnostica per immagini nella patologia degenerative del sistema extra-piramidale. Radiol Med 106:19-23.
- Nakagawa N, Katayama T, Makita Y, Kuroda K, Aizawa H (1999) A case of spinocerebellar ataxia type 6 mimicking olivopontocerebellar atrophy. Neuroradiology 41: 505-503. [Crossref]
- Satoh JI, Tokumoto H, Yukitake M (1998) Spinocerebellar ataxia type 6: MRI of three Japanese patients. Neuroradiology 40: 222-227.
- Nakayama T, Nakayama K, Takahashi Y, (2001) Case of spinocerebellar ataxia type 1 showing high intensity lesions in the frontal white matter on T2-weighted magnetic resonance images. Med Sci Monit 72: 299-303.
- Klockgether T, Skalej M, Wedekind D (1998) Autosomal dominant cerebella ataxia type I MRI-based volumetry of posterior fossa structures and basal ganglia in spinocerebellar ataxia types 1, 2 and 3. Brain 121: 1687-1693.
- Hearn E, Holmes SE, Calvert PC, Ross CA, Margolis RL (2001) SCA- 12: tremor with cerebellar and cortical atrophy is associated with a CAG repeat expansion. Neurology 56: 299-303.
- Day JW, Schut LJ, Moseley ML, Durand AC, Ranum LP (2000) Spinocerebellar ataxia type 8. Neurology 55: 649-657. [Crossref]
- Ikeda Y, Shizuka M, Watanabe M, Schmitt M, Okamoto K (2000) Asymptomatic CTG expansion at the SCA8 locus is associated with cerebellar atrophy on MRI. J Neurol Sci 182: 76-79. [Crossref]
- Stevanin G, Herman A, Brice A, Du¨rr A (1999) Clinical and MRI findings in spino-cerebellar ataxia type 5. Neurology 53: 1355-1357. [Crossref]
- Giuffrida S, Saponara R, Restivo D (1999) Supratentorial atrophy in spinocerebellar ataxia type 2: MRI study of 20 patients. J Neurol 246: 383-388. [Crossref]
- Oka M, Katayama S, Imon Y, Ohshita T, Mimori Y (2001) Abnormal signals in proton density-weighted MRI of the superior cerebellar peduncle in progressive supranuclear palsy. Acta Neurol Sc 104:1-5.
- Macia F, Yekhlef F, Ballan G (2001) T2 hyperintense lateral rim and hypointense putamen are typical but not exclusive of multiple system atrophy. Arch Neurol 58: 1024-1025.
- Leehey MA (2009) Fragile X-associated tremor/ataxia syndrome: clinical phenotype, diagnosis, and treatment. J Investig Med 57: 830-836.
- Ormerod IE, Miller DH, Donald WI, Boulay EP, Rudge P, et al. (1987) The role of NMR imaging in the assessment of multiple sclerosis and isolated neurological lesions. A quantitative study. Brain 110: 1579-1616.
- Gonçalves FG, Lamb L (2011) Progressive multifocal leukoencephalopathy restricted to the posterior fossa in a patient with systemic lupus erythematosus. Braz J Infect Dis15: 609-612.
- Horger M, Beschorner R, Beck R, Nägele T, Schulze M (2012) Common and uncommon imaging findings in progressive multifocal leukoencephalopathy (PML) with differential diagnostic considerations. Clin Neurol Neurosurg 114: 1123-1130.
- Bartynski WS, Boardman JF (2007) Distinct imaging patterns and lesion distribution in posterior reversible encephalopathy syndrome. Am J Neuroradiol 28: 1320-1327.
- McKinney AM, Jagadeesan BD, Truwit CL (2013) Central-variant posterior reversible encephalopathy syndrome: brainstem or basal ganglia involvement lacking cortical or subcortical cerebral edema. Am J Roentgenol 201: 631-638.
- Morales H, Tomsick T (2015) Middle cerebellar peduncles: Magnetic resonance imaging and pathophysiologic correlate. World J Radiol 28: 438-47.
- Geibprasert S, Gallucci M, Krings T (2010) Addictive illegal drugs: structural neuroimaging. Am J Neuroradiol 31: 803-808.
- Okamoto K, Tokiguchi S, Furusawa T, Ishikawa K, Quardery AF, et al. (2003) MR features of diseases involving bilateral middle cerebellar peduncles. Am J Neuroradiol 24: 1946-1954.
- Uchino A, Sawada A, Takase Y, Kudo S (2004) Symmetrical lesions of the middle cerebellar peduncle: MR imaging and differential diagnosis. Magn Reson Med Sci 3: 133-140.
- Arora A, Neema M, Stankiewicz J, Guss ZD, Guss JG, et al. (2008) Neuroimaging of toxic and metabolic disorders. Semin Neuro 28: 495-510. [Crossref]
- Kuhn MJ, Johnson KA, Davis KR (1988) Wallerian degeneration: evaluation with MR imaging. Radiol 168: 199-202.
- Castillo M, Mukherji SK (1999) Early abnormalities related to postinfarction Wallerian degeneration: evaluation with MR diffusion-weighted imaging. J Comput Assist Tomogr 23: 1004-1007.
- Figatowska M, Duczkowski M, Madzik J, Uliasz M, Zawadka A (2012) Diffusion-Weighted Imaging of the Early Phase of Wallerian Degeneration. A Report of two Pediatric Cases and Literature Review. Neuroradiol J 25: 657-664.
- Simone T, Gladin C, Carriero MR, Farina L (2005) Wallerian degeneration of the pontocerebellar fibers. Am J Neuroradiol 26: 1062-1065.
- Fitzek C, Fitzek S, Stoeter P (2004) Bilateral Wallerian degeneration of the medial cerebellar peduncles after ponto-mesencephalic infarction. Eur J Radiol 49: 198-203.
- Küker W, Schmidt F, Heckl S, Nägele T, Herrlinger U (2004) Bilateral Wallerian degeneration of the middle cerebellar peduncles due to paramedian pontine infarction: MRI findings. Neuroradiology 46: 896-899.
- Gala F, Becker A, Pfeiffer M, Kollias S (2013) Acute Wallerian degeneration of middle cerebellar peduncles due to basilar artery thrombosis. Indian J Radiol Imaging 23: 164-167.
- Ellison D, Love S, Chimelli L, Harding BN, Lowe J (2004) Neuropathology: A Reference Text of CNS Pathology (2nd edition). MOSBY, Elsevier Ltd p. 879.