TSLC1, HTLV-1, KSHV, NF-κB, NFAT, vFLIP, vGPCR, Tax
A vast number of cell surface molecules (CSMs), which are involved in the regulation of inflammation, innate and adaptive immune response, cell death, and the restoration of tissue homeostasis were recently identified. Whereas some of these CSMs serve the role of cell adhesive molecules, others function as costimulatory molecules to unlock distinct inflammatory and immune responses [1,2]. In addition, certain CSM heterophilic and homophilic interactions serve the role of either ligands or receptors, or both in this process [3]. CSMs also play a critical role in cell-to-cell communication [4]. The fate of the tissue is determined by the levels of CSMs expressed in a particular tissue. Certain levels of CSM expression are sufficient for their beneficial function; however, altering levels of CSM expression due to infection, injury, or substance abuse play a detrimental role in a particular tissue. Interestingly, the mechanisms of CSM-mediated distinct effects on regulation of inflammation, innate and adaptive immunity, and the optimal levels of CSM expression required for beneficial effects are poorly understood. This review will focus on recent findings on the mechanism of inflammation regulation mediated by a highly upregulated CSM, tumor suppressor in lung cancer-1 (TSLC1), in oncogenic virus infected cells.
Approximately 12-20% of all human cancers worldwide are caused by infections with oncogenic viruses. For example, Human T-cell leukemia virus type 1 (HTLV-1), Κaposi's sarcoma herpesvirus/human herpesvirus 8 (KSHV/HHV8), Epstein Barr virus (EBV), hepatitis B virus (HBV), hepatitis C virus (HCV), and human papillomavirus (HPV), are oncogenic viral agents responsible for a large number of human cancers. This review will mainly focus on a CSM-TSLC1 involved in KSHV and HTLV-1 oncogenesis. KSHV is one of the oncogenic viruses responsible for human cancers, including Kaposi’s sarcoma (KS), Primary Effusion Lymphoma (PEL), and the lymphoproliferative disorder Multicentric Castleman’s disease (MCD). Chronic inflammation mediated by KSHV infection plays a decisive role in the development and survival of cancer cells. HTLV-1 is a retrovirus and etiological agent of adult T-cell leukemia (ATL). These viruses encode unique sets of viral genes that regulate viral replication and promote distinct malignancies. ΚSHV is a DNA virus and encodes several oncogenes, such as KSHV-encoded chemokine receptor or viral G protein-coupled receptor (vGPCR) and antiapoptotic viral Fas-associated death domain-like interleukin-1β-converting enzyme-inhibitory protein (vFLIP). HTLV-1 encodes the Tax trans-activating protein and oncogene. The KSHV-encoded oncogenes vGPCR and vFLIP are key KSHV lytic and latent genes, respectively. Viral oncogenes, vFLIP of KSHV and Tax of HTLV-1 are required for viral transformation, cell proliferation, cell survival and genetic instability. Our published work and preliminary data suggest that TSLC1 is highly induced during KSHV and HTLV-1 de novo infection and is critical for the establishment of KSHV and HTLV-1 latency. In addition, viral oncogenes, vFLIP, vGPCR, and Tax, dysregulate the tumor suppressor function of TSLC1 and hijack it’s function to maintain chronic inflammation required for the KSHV and HTLV-1-induced tumors. TSLC1 expression is critical for the survival of KSHV and HTLV-1-infected tumor cells. However, the mechanisms of viral oncogene-mediated dysregulation of TSLC1 function are poorly understood. TSLC1 is not only expressed on the cell surface but also localized in the cytoplasm and other cell organelles, such as the endoplasmic reticulum and Golgi bodies. Therefore, it will be of great interest for future studies to determine whether the high levels of TSLC1 expressed in KSHV and HTLV-1 infected cells are also involved in virus endocytosis or exocytosis, which could potentially be targeted by small molecules to limit the de novo infection and spread of these oncogenic viruses.
Chronic inflammation mediated by transcription factors including, Nuclear factor-kappaB (NF-κB), Signal transducer and activator of transcription 3 (STAT3), and Nuclear factor of activated T-cells (NFAT), in oncogenic virus-infected cells play critical roles in viral transformation, cell proliferation, cell survival and genetic instability [5]. KSHV and HTLV-1 often establish a life-long asymptomatic latent infection [5]. Infections with ΚSHV and HTLV-1 can immortalize human primary endothelial cells and B cells, or T cells, respectively [6]. Chronically activated NF-κB pathways mediated by vFLIP and vGPCR of KSHV, or Tax of HTLV-1 are critical for the survival of ΚSHV and HTLV-1 infected cells [7,8]. We and others have shown that TSLC1 is highly upregulated in ΚSHV and HTLV-1-infected cells. Interestingly, TSLC1 is a critical molecule for vFLIP, vGPCR and Tax-mediated chronic activation of the IKK kinase complex (IKK⍺ IKKβ, and IKKɣ), which is required for the phosphorylation and proteasomal degradation of the NF-κB inhibitor, IκB⍺. Viral oncogene-mediated degradation of IκB⍺ induces the activation of NF-κB dimers (e.g. p50/p65), which maintains upregulation of several hundred genes, including pro-inflammatory cytokines and chemokines in KSHV and HTLV-1 infected cells (Figure 1) [7,8]. NFAT is chronically activated by HTLV-1 Tax and KSHV vGPCR [7,9]. A recent study suggests that TSLC1 is also critical for vGPCR-mediated chronic activation of NFAT [7]. Although STAT3 is chronically activated in KSHV and HTLV-1-infected tumor cells, it is unknown if TSLC1 is also involved in chronic activation of STAT3 in these virus infected tumor cells. Since TSLC1 is critical for the survival of KSHV and HTLV-1 infected tumor cells, we speculate that TSLC1 may also regulate STAT3 activation in these virus infected cells. Previous studies have shown that several GPCRs activate STAT3 and NF-κB, which contributes to the development of cancers. Although our published results and preliminary data suggest that an interaction of TSLC1 with vGPCR is critical for the activation of NF-κB, it is unknown whether TSLC1 also interacts with host GPCRs, which are involved in the activation of NFAT, STAT3 and NF-κB. Therefore, it will be interesting in future studies to determine whether TSLC1 is also involved in activation of STAT3 and NF-κB in uninfected cells/tissues as part of its normal physiological role.
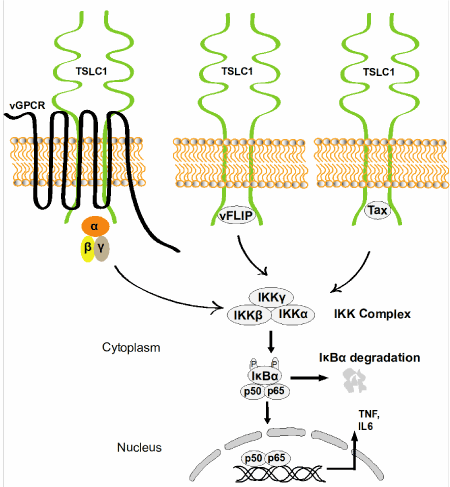
Figure 1. Chronic NF-κB activation by KSHV and HTLV-1 oncogenes. TSLC1 localized at the plasma membrane mediates viral oncogene (vGPCR, vFLIP, and Tax)-induced chronic activation of NF-κB (p50/p65) and inflammation in KSHV and HTLV-1 infected cells.
The authors have declared that no competing interests exist.
This research was funded by American Cancer Society RSG-16-254-01-MPC. We gratefully acknowledge Dr. Edward Harhaj for review of this manuscript.
- Brubaker SW, Bonham KS, Zanoni I, Kagan JC (2015) Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol 33: 257-290. [Crossref]
- Zitvogel L, Kepp O, Kroemer G (2010) Decoding cell death signals in inflammation and immunity. Cell 140: 798-804. [Crossref]
- Duraivelan K, Basak AJ, Ghosh A, Samanta D (2018) Molecular and structural bases of interaction between extracellular domains of nectin-2 and N-cadherin. Proteins 86: 1157-1164. [Crossref]
- Guo F, Peng Li, French JB, Mao Z, Zhao H, et al. (2015) Controlling cell-cell interactions using surface acoustic waves. Proc Natl Acad Sci U S A 112: 43-48.
- Purushothaman P, Uppal T, Sarkar R, Verma SC (2016) KSHV-Mediated Angiogenesis in Tumor Progression. Viruses 8: 7 [Crossref]
- Pique C, Jones KS (2012) Pathways of cell-cell transmission of HTLV-1. Front Microbiol 3: 378 [Crossref]
- Hunte R, Alonso P, Thomas R, Bazile CA, Ramos JC, et al. (2018) CADM1 is essential for KSHV-encoded vGPCR-and vFLIP-mediated chronic NF-kappaB activation. PLoS Pathog 14: e1006968. [Crossref]
- Pujari R, Hunte R, Thomas R, van der Weyden L, Rauch D, et al. (2015) Human T-cell leukemia virus type 1 (HTLV-1) tax requires CADM1/TSLC1 for inactivation of the NF-kappaB inhibitor A20 and constitutive NF-kappaB signaling. PLoS Pathog 11: e1004721. [Crossref]
- Zhang J, He S, Wang Y, Brulois K, Lan K, et al. (2015) Herpesviral G protein-coupled receptors activate NFAT to induce tumor formation via inhibiting the SERCA calcium ATPase. PLoS Pathog 11: e1004768. [Crossref]