The aim of the present investigation was to evaluate the role of carnosine on aging-induced Aβ deposition in different brain regions (prefrontal cortex, hippocampal CA1 and CA3 zones, hypothalamus, pons-medulla and cerebellum) in connection with the locomotor activity (open field locomotion and rearing count) and corticosterone status in plasma and adrenal gland. During aging, both in 18 and 24 months aged rats (a) a graded deposition of Aβ in their brain regions was observed with a maximum deposition in prefrontal cortex of 24 months aged rats without any Aβ deposition in cerebellum, (b) the plasma and adrenal corticosterone level was increased, and (c) locomotor activity were reduced. Carnosine (2.0 µg/Kg/day, i.t.; for 21 consecutive days) reduced the aging-induced brain regional graded Aβ deposition and attenuated the aging-induced increase of both plasma and adrenal corticosterone level as well as the locomotor activity. Thus, it may be concluded from these observations that (a) aging reduces both rearing and locomotor activities which may be correlated with the increase of corticosterone in both plasma and adrenal gland with a graded Aβ accumulation in the brain regions, except cerebellum and (b) carnosine attenuates these aging-induced phenomena, observed in those brain regions, plasma and adrenal gland, and also in locomotor activities.
aging, brain regions, amyloid-beta, corticosterone, locomotor activity, carnosine
The aging process of living species makes the phenomena unique as its negative association with the ability to respond to stress-related physiological conditions [1]. This leds to diverse deleterious changes in cells and tissues, and also creates a positive consequence to culminate into the risk of aging-induced diseases towards death [2]. It’s long been known that the physical or psychological stress factors can trigger up the activity of the hypothalamic-pituitary-adrenal (HPA) axis which in turn may increase corticosterone level. The glucocorticoids are found in the rodents’ blood as corticosterone, and in humans as cortisol [3]. Corticosterone releases from the adrenal cortex in response to the release of adrenocorticotropic hormone (ACTH) into the bloodstream from hypothalamus. Its excess presence in plasma induces stress by activating different enzymes which produce some precursors of ROS (reactive oxygen species) and/or RNS (reactive nitrogen species) as their end products (e.g., H2O2 from monoamine oxidase-A activity) often influence functions of the brain regions [4,5]. The stressors trigger the HPA axis by the activation of parvocellular neurons of the paraventricular nucleus (PVN) of hypothalamus [2]. The release of corticosterone is regulated by the negative feedback mechanism in which corticosterone binds to receptor which resides in pituitary and PVN [2]. Glucocorticoid receptors involved in the recovery from stress with an activation of a set of hormonal and neuronal responses and other mediators, such as neurotransmitters, cytokines to assist in adaptation to a new situation or challenge [6]. This corticosterone has been found to be associated with the amyloid pathogenesis [7] and loss of neural integrity and functions of brain regions [8]. Interestingly, weight of human brain and/or its volume has (have) been found to decline during aging [9] with production of a wide range of misfolded proteins, aggregation (proteinopathy) and loss of neuronal cells [10]. It is well-known that deposition and formation of brain regional synaptic plaques are the major histopathological hallmark of brain aging [10]. In different aging-induced neurodegenerative diseases, the senile plaque formation due to Aβ peptide deposition has been claimed to be the signs of mutation of genes coded for amyloid precursor protein (APP) or presenilins [11]. In consequence of these gene mutations during aging, various neurobiochemical alterations such as stress related overproduction of corticosterone, ROS and RNS have a reflection on Aβ peptide which makes the scenario of synaptic senile plaque formation more deleterious [7,10]. Previously, Fukumoto et al. [12] have shown that the senile plaque has been found not only in aging-induced neurodegenerative diseases but also in the non-diseased aging pathology with the same characteristics of Aβ protein deposition in different brain regions. Interestingly, it has also been observed that the aging-induced changes do not occur homogeneously in all the brain regions [4,9,13,14]; but, region-specific Aβ deposition in relation to the plasma and adrenal corticosterone during aging has not yet been well understood. The adrenal gland being the site of synthesis of corticosterone demands an attention for further study in relation to the plasma corticosterone level and brain regional Aβ deposition for plaque formation during aging.
The increase in ROS with the reduction in the antioxidant system during aging develops the antioxidant-ROS imbalance and the application of different antioxidant molecules to the elderly individuals to prevent or delay the aging process is one of the promising fields of geriatric research [15]. Among the antioxidant molecules, carnosine is one of them. Carnosine, an endogenous dipeptide (β-Ala-L-His) biomolecule having antioxidant and antiglycating properties is present in blood, brain and muscles [13,16,17]. This biomolecule is synthesized by carnosine synthase in presence of a rate-limiting amino acid, β-alanine [18] and is degraded by carnosinase [19]. Carnosine has gene regulatory property [17], anti-senescence activity and has been reported to inhibit metastasis [17]. Margles [20] has also shown that olfactory bulb is an enriched zone of carnosine and smelling sense is lost (hyposmia) with a loss of carnosine concentration in aging-induced diseases. In oxidative and nitrosative driven neuro diseases carnosine has been found to act as a potent neuroprotectant by scavenging the reactive oxygen species [16,17]. Since carnosine has (a) these unique properties, and (b) ability to attenuate the aging-induced brain regional deterioration in serotonergic activity [13,21], without any side effect [22], it is not unreasonable to assume that there may be a role of carnosine on aging-induced stress related to corticosterone status and brain regional Aβ deposition. Based on this correlative information, the present study has focused with the effect of carnosine on aging- should be before induced changes in the plasma and adrenal corticosterone status as well as locomotor behaviour, and brain regional Aβ (1-42) levels including plaques.
Materials
L-carnosine and poly-L-lysine were purchased from Sigma Chemicals (St. Louis, MO, USA). Other chemicals including chloroform, sodium hydroxide, sodium chloride, sulfuric acid were purchased from Merck-India (Worli-Mumbi), India. EXPOSE mouse and rabbit specific HRP/DAB detection IHC kit (ab80436) and rabbit polyclonal to β-amyloid 1-42 (ab10148) were purchased from Abcam (Cambridge, U.K.). Ethyl alcohol (EtOH) was purchased from Bengal Chemical (Kolkata, India), ELISA kit (KMB3441) from Invitrogen Corporation (California, U.S.A.).
Animals and animal care: Wistar strain male albino rats were used in the present study. The rats were kept in a room having a 12 h light/ 12 h dark cycle with a constant relative humidity (70±5%) and temperature 25±0.5°C. Animals were maintained with a supplementation of normal standard laboratory diet and water ad libitum. The Institutional Animal Ethical Committee (Jadavpur University) has approved the protocol of the present study and all efforts were made to minimize the number of animals used and their sufferings, if any.
Experimental design: The rats of the present study were divided into three different age groups (4 months young: group 1; 18 months aged: group 2; and 24 months aged: group 3) and each group were subdivided into three subgroups (subgroups 1a, 1b, 1c; subgroups 2a, 2b, 2c and subgroups 3a, 3b, 3c respectively). Each subgroup contained 4-6 rats (Figure 1). The animals of the subgroups 1a, 2a and 3a were considered as without vehicle-treated or control subgroup. The subgroups 1b, 2b, 3b were treated with a volume (20 µl) of vehicle (saline) without carnosine through the same route under similar condition for 21 consecutive days and were considered as control rats of the corresponding experimental (carnosine treated) subgroups (subgroups 1c, 2c, 3c). The subgroups 1c, 2c and 3c were treated with carnosine in 20 µl saline 2.0 µg/kg/day intrathecally (i.t.) for 21 consecutive days. The above-mentioned age groups (4, 18 and 24 months) of rats were attained their desired age after completion of the duration (21 consecutive days) of treatment. The control, vehicle-treated and experimental groups of animals was sacrificed after 4 h of the last administration of carnosine or its vehicle between 09:00 am-10:00 am to avoid the circadian effect, if any.
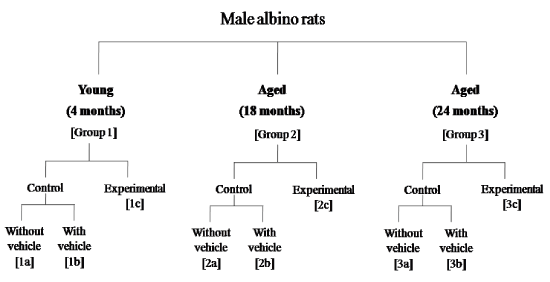
Figure 1. Schematic diagram of the experimental design
Collection of plasma and adrenal gland: Immediately after sacrifice of rats of both experimental and their corresponding control groups, the blood was collected and isolated the plasma immediately, following the method of Banerjee and Poddar [23] and stored at 4 ̊ C for further assay. The adrenal glands were isolated from kidneys and freed from adhering tissues before weighing and collected into the 0.9% NaCl-EtOH solution (1:4) for further analysis.
Estimation of corticosterone from plasma and adrenal gland: The corticosterone level of plasma and adrenal gland was measured by spectrofluorometer following the method of Bandyopadhyay and Poddar [24]. Briefly, the plasma was isolated from whole blood by centrifugation at 2500 rpm for 10 min at 4 ̊ C and the adrenal glands were homogenized and centrifuged at 4000 rpm for 10 min at 4 ̊ C. The plasma was diluted 10 times. The chloroform was added 7.5 times into both the diluted plasma and supernatant of homogenized adrenal gland separately, mixed well and centrifuged (2500 rpm for 5 min at 4 ̊ C). 0.1N NaOH solution was added into the organic layer, mixed well and centrifuged (2500 rpm for 5 min at 4 ̊ C). The fluorescent reagent (contains concentrated sulphuric acid and ethyl alcohol) was added into the organic layer, mixed well and centrifuged (2500 rpm for 5 min at 4 ̊ C). The fluorescence of those organic layers was measured by spectrofluorometer (Perkin Elmer LS55) with the λex 470 nm and λem 520 nm. The corticosterone level was expressed as µg/mg protein in the adrenal gland and µg/100 ml in plasma.
Collection of brain tissue: Immediately after sacrifice of rats of both experimental and their corresponding control groups, the brains were collected and (a) immediately sectioned under frozen (-20˚C) condition using cryostat microtome (Leica CM 1850) for the Immunohistochemical (IHC) staining, (b) were dissected out of different brain regions (prefrontal cortex, hippocampus, hypothalamus, pons-medulla and cerebellum) under cold condition (0-4˚C) according to the method as described by Poddar and Dewey [25] for quantification of Aβ by ELISA.
Neuronal plaques identification and quantification: The presence of neuronal plaques in different brain regions [prefrontal cortex, hippocampus (CA1 and CA3), hypothalamus, pons-medulla and cerebellum] of both control and experimental groups of young (4 months) and aged (18 and 24 months) rats were identified qualitatively and quantitatively using immunohistochemical (IHC) staining with anti-β-amyloid antibody according to the protocol provided by Abcam Biotechnology Company (Cambridge, UK). All the IHC-stained coronal sections were analyzed manually with 20 X magnification using a binocular compound microscope (Olympus BX 51, Cool SNAP cf color/OL).
Immunohistochemical (IHC) staining: The rat brains were used for the IHC staining with rabbit polyclonal β-Amyloid 1-42 antibody, EXPOSE mouse and rabbit specific HRP/DAB detection IHC kit according to the instruction given by Abcam. In brief, frozen sections with 10 μm thickness were taken in poly-L-lysine coated glass slides. Sections were at first covered with hydrogen peroxide block to reduce endogenous peroxidase action. Then 0.025% Triton X-100 in tris-buffer saline (TBS) was applied followed by protein block. Sections were then incubated with rabbit polyclonal β-amyloid 1-42 antibody at 4˚C for overnight. After incubation, the sections were washed with TBS and treated sequentially with complement, HRP conjugate and DAB chromogen. After washing with TBS, sections were counterstained by haematoxylin, dehydrated with graded alcohol, cleared in xylene and mounted with DPX [26].
Estimation of brain regional Aβ (1-42) steady state level: The brain regional Aβ (1-42) steady state level was measured by ELISA kit, according to the protocol provided by Invitrogen Corporation (California, USA). Briefly, the brain regions mentioned above were collected after dissection into 4X PBS (phosphate buffer solution) with protease inhibitor cocktail and homogenized with a continuous addition of guanidine-HCl/Tris-HCl solution, centrifuged (16000 g, for 20 min, at 4 ̊ C) and supernatant was collected for ELISA (vide suppliers protocol). The absorbance was measured at 450 nm in ELISA microplate reader (Bio-Rad, California, USA). The brain regional Aβ (1-42) steady state level was expressed as pg/mg protein. Protein content of tissue was estimated spectrophotometrically following the method of Lowry et al. [27] using bovine serum albumin (BSA) as standard.
Assessment of locomotor activity: The locomotor activity was assessed with the measurement of (a) vertical (rearing) motor activity, and (b) open field locomotion together. The rearing count (times/5 min) was measured by the method as described by Dalal and Poddar [28] and open field locomotion (times/ 10 min) following the method as described by Turner and Burne [29]. In brief, (a) the animals were gently placed from their home cages into the transparent plastic closed chamber (24×24×20 cm3) and the vertical lid was closed. An electrical lamp was placed at the top of the chamber. The vertical rearing movement frequencies of rats were observed and quantified for 5 min by an electrical device and also manually. The rats were placed back again in their own home cages after the routine study; (b) the rats were placed at the centre of the horizontal open field arena (60×60×45 cm). The arena was divided equally with 25 squares. The locomotion of rats was quantified with the number of square crossed in 10 min time period. Upon completion of the time period the rats were kept in their respective home cages.
Statistical analysis: Statistical analyses of the quantitative data were assessed by the analyses of variance (ANOVA) with a post hoc Tukey’s test unless otherwise mentioned. p<0.05 was considered as level of significance.
Corticosterone levels in plasma and adrenal gland during aging: In 18 and 24 months aged rats the level of corticosterone was found to increase significantly in (a) plasma (37.73%, p<0.001 and 129.07%, p<0.001 respectively) and (b) adrenal gland (60.00%, p<0.001 and 86.92%, p<0.001 respectively) with respect to the values obtained in young (4 months) rats treated with vehicle (Table 1).
Table 1. Effect of carnosine on the steady state level of corticosterone during aging in male (albino rats Results
Site of study |
Vehicle treated rats |
Carnosine treated (2.0µg/Kg/day, i.t.; for 21 consecutive days) rats |
4 months |
18 months |
24 months |
4 months |
18 months |
24 months |
Adrenal gland (µg/mg protein) |
100.00
±3.01 |
160.00
±9.17a |
186.92
±8.36a |
92.31
±12.12 |
150.00
±15.47 |
90.00
±8.26b |
Plasma
(µg/100 ml) |
100.00
±2.31 |
137.73
±15.69a |
229.07
±24.22a |
164.72
±15.32a |
150.55
±17.64 |
166.80
±16.18b |
(percent) are expressed as mean±SEM of 4 separate observations. Each observation was made from a single rat. The vehicle treated corticosterone level of (a) adrenal gland (21.60 µg/mg protein) and (b) plasma (1.30 µg/100ml) in young 4 months rats respectively were considered as 100.00. There was no significant difference between results of the vehicle treated rats and the rats of without vehicle treatment. Significantly different from corresponding vehicle treated (a) 4 months rats ap<0.001, (b) 24 months rats bp<0.001
Aging-induced brain regional Aβ (1-42) level: Aβ (1-42) levels in aged (18 months and 24 months) rats were increased significantly in prefrontal cortex (63.36%, p<0.001 and 104.96%, p<0.001 respectively), hippocampus (20.29%, p<0.05 and 121.34%, p<0.001 respectively), hypothalamus (28.03%, p<0.01 and 140.97%, p<0.001 respectively) and pons-medulla (22.92%, p<0.05 and 93.75%, p<0.001 respectively) without any significant change in its cerebellum with respect to the corresponding data obtained in young (4 months) rats (Table 2). The prefrontal cortex, among the brain regions showed the highest Aβ (1-42) level in 18 months aged rats whereas, with the progression of age, in 24 months aged rats the hypothalamus showed the highest level of Aβ (1-42) followed by hippocampus> prefrontal cortex> pons-medulla. Though, in young rats (4 months) the basal level of Aβ (1-42) was found to be highest in cerebellum, followed by hippocampus, hypothalamus, prefrontal cortex and pons-medulla (Table 2).
Table 2. Steady state level of amyloid beta (1-42) in different brain regions of young and aged rats with or without carnosine treatment
Age groups |
Age (months) of animals |
Brain regional amyloid beta (1-42) content (pg / mg protein) |
Prefrontal cortex |
Hippocampus |
Hypothalamus |
Pons-medulla |
Cerebellum |
Vehicle treated |
Carnosine treated |
Vehicle treated |
Carnosine treated |
Vehicle treated |
Carnosine treated |
Vehicle treated |
Carnosine treated |
Vehicle treated |
Carnosine treated |
Young |
4 |
1.82
±0.145 |
1.76
±0.134 |
2.39
±0.19 |
2.32
±0.175 |
1.86
±0.185 |
1.95
±0.165 |
0.48
±0.045 |
0.58
±0.05 |
4.87
±0.295 |
5.16
±0.315 |
Aged |
18 |
2.97
±0.165a |
1.61
±0.119d |
2.88
±0.215 |
2.22
±0.180e |
2.38
±0.185b |
2.59
±0.224 |
0.59
±0.045c |
0.47
±0.045 |
5.63
±0.315 |
3.52
±0.249d |
24 |
3.72
±0.19a |
1.83
±0.15f |
5.29
±0.260a |
1.97
±0.190f |
4.47
±0.240a |
2.40
±0.180f |
0.93
±0.075a |
0.42
±0.044f |
4.99
±0.275 |
4.76
±0.298 |
(Results are expressed as mean±SEM of 4-6 separarte observations. Each observation was made from a single rat. No significant difference was observed between the results of vehicle treated and without vehicle treated groups of rats. Significantly different from the corresponding vehicle treated (a) young rats ap<0.001, bp<0.01, cp<0.05; (b) age-matched (i) 18 months dp<0.001, ep<0.05, (ii) 24 months fp<0.001)
Presence of Aβ plaques in brain regions during aging: Figures 2-5 represent that the presence of brain regional Aβ (1-42) plaques was much higher, both qualitatively and quantitatively in the 24 months aged rat brain regions than that observed in the corresponding brain regions of 18 months aged rats. It is further evident from the data of Figures 2a-5a that in 24 months aged rats the number of Aβ plaque (per 107 µm2 area) significantly increased in prefrontal cortex (39.20%, p<0.01), hippocampal CA1 zone (36.98%, p<0.01), hippocampal CA3 zone (35.55%, p<0.01), hypothalamus (29.75%, p<0.01) and pons-medulla (24.95%, p<0.01) compared to that found in 18 months aged rats. No Aβ plaques were found in any of the brain regions studied in 4 months young rats (Figures 2-6). In cerebellum neither in young (4 months) nor in aged (18 and 24 months) rats, Aβ plaque was seen Figures 6.
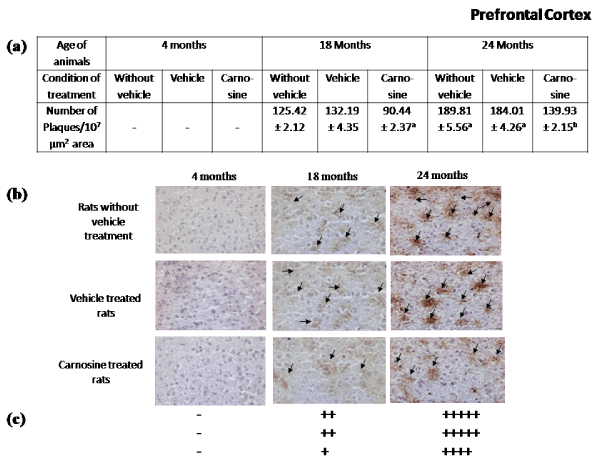
Figure 2. Effect of carnosine on the presence of Aβ plaque in the prefrontal cortex of young (4 months) and aged (18 and 24 months) male albino rats.
(a) Represents number of Aβ plaque during aging (18 and 24 months). Results are expressed as mean±SEM of 4-6 separate observations.
Significantly different from corresponding control (vehicle treated) aged (i) 18 months rats ap<0.01and (ii) 24 months rats bp<0.01; No statistical significant difference was observed between the results of the rats (irrespective of the age groups) with or without the vehicle of carnosine treated group.
(b) represents results of the tissue sections using immunohistochemical staining, and
(c) represents the qualitative presentation of the results (b).
Black arrow heads indicate the Aβ plaques. The tissue sections were observed under the bright field compound microscope with 20X magnification. +++++ indicates extreme high, ++++ indicates high, +++ indicates medium, ++ indicates low, + indicates very low, ̶ indicates absence of Aβ plaque
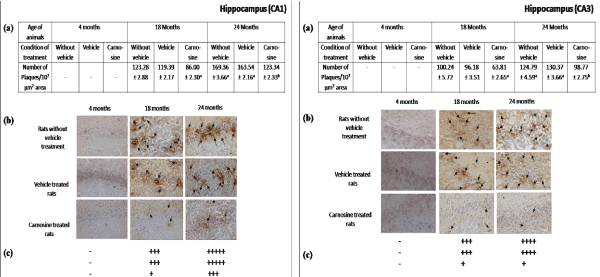
Figure 3. Effect of carnosine on the presence of Aβ plaque in the hippocampus (CA1 and CA3) of young (4 months) and aged (18 and 24 months) male albino rats
(a) Represents number of Aβ plaque during aging (18 and 24 months). Results are expressed as mean±SEM of 4-6 separate observations.
Significantly different from corresponding control (vehicle treated) aged (i) 18 months rats ap<0.01and (ii) 24 months rats bp<0.01. All other details are same as described in Figure 2
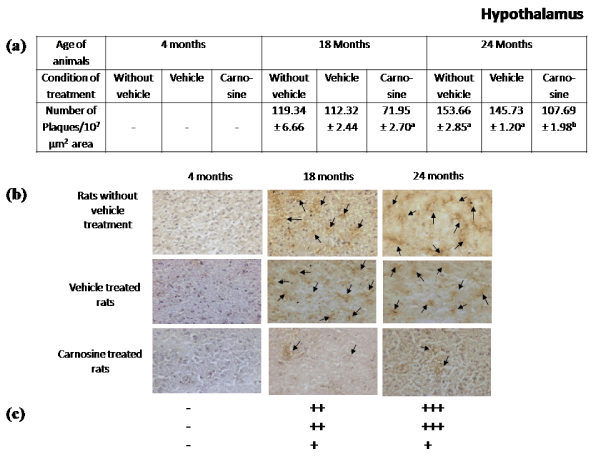
Figure 4. Effect of carnosine on the presence of Aβ plaque in the hypothalamus of young (4 months) and aged (18 and 24 months) male albino rats
(a) Represents number of Aβ plaque during aging (18 and 24 months). Results are presented as mean±SEM of 4-6 separate observations.
Significantly different from corresponding control (vehicle treated) aged (i) 18 months rats ap<0.01and (ii) 24 months rats bp<0.01. All other details are same as described in Figure 2
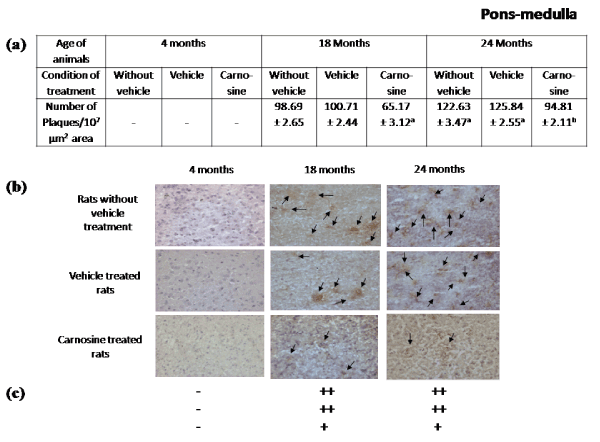
Figure 5. Effect of carnosine on the presence of Aβ plaque in the pons-medulla of young (4 months) and aged (18 and 24 months) male albino rats. (a) Represents number of Aβ plaque during aging (18 and 24 months). Results are presented as mean±SEM of 4-6 separate observations
Significantly different from corresponding control (vehicle treated) aged (i) 18 months rats ap<0.01and (ii) 24 months rats bp<0.01. All other details are same as described in Figure 2
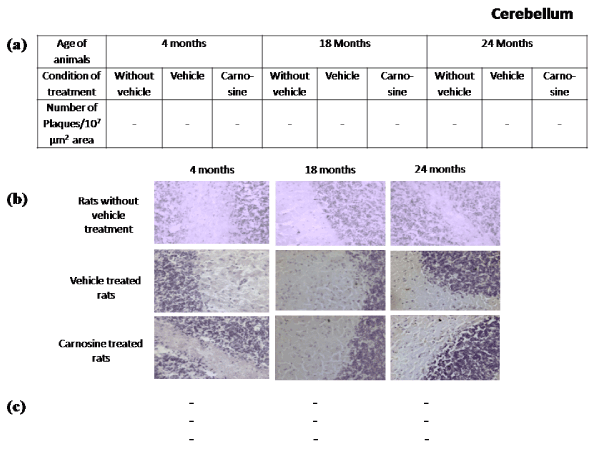
Figure 6. Effect of carnosine on the presence of Aβ plaque in the cerebellum of young (4 months) and aged (18 and 24 months) male albino rats
(a) Represents the Aβ plaque number during aging (18 and 24 months). No plaque was found in this brain region. All other details are same as described in Figure 2
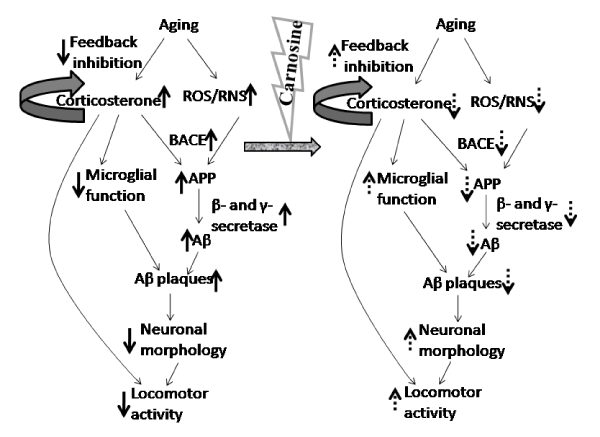
Figure 7. Mechanistic overview of the role of carnosine on aging-induced plasma corticosterone and brain regional Aβ plaque deposition in relation to locomotor activity indicates aging-induced increase; indicates aging-induced decrease; indicates feedback mechanism; indicate carnosine-induced attenuation of aging-induced increase and decrease respectively
Aging-induced rearing count and open field locomotion activity study: The rearing count, and open field locomotion activity, presented in Table 3 were found to decrease during aging, and this (decrease) was more in 24 months aged (72.73%, p<0.001 and 70.73%, p<0.001 respectively) rats than those observed in 18 months aged (31.82%, p<0.001 and 30.08%, p<0.001 respectively) rats in comparison to those data obtained from 4 months young rats treated with vehicle alone.
Table 3. Effect of carnosine on Locomotor activity (times/5 mins) during aging in male albino rats Results
Conditions of treatment |
Locomotor activity |
Rearing count (times/5 min) |
Open field locomotion (times/10 min) |
Young
(4 months) |
Aged
(18 months) |
Aged
(24 months) |
Young
(4 months) |
Aged
(18 months) |
Aged
(24 months) |
Vehicle treated |
100.00
±18.18 |
68.18
±13.64a |
27.27
±9.09a |
100.00
±9.76 |
69.92
±7.32a |
29.27
±6.50a |
Carnosine treated |
109.09
±13.64 |
81.82
±13.64 |
72.73
±13.64b |
114.63
±12.19 |
79.67
±8.94 |
62.60
±8.13b |
(percent) are expressed as mean±SEM of 4-6 separate observations. Each observation as made from a single rat. No significant difference was observed between the results of (a) rearing count, and (b) open field locomotion with and without vehicle of carnosine treated rats. The control values of rearing count (23.25±1.44) and open field locomotion count (116.37±8.70) in young 4 months vehicle treated rats were considered as 100.00. Significantly different from the corresponding control (vehicle treated) (i) 4 months young rats ap<0.001, (ii) age-matched aged (18 and 24 months) rats bp<0.001)
Effect of carnosine on aging-induced increase of plasma and adrenal corticosterone level: This is evident in Table 1 that the treatment of carnosine (2.0 µg/Kg/day, i.t.) for 21 consecutive days significantly attenuated the aging-induced increase of (a) plasma (27.18%, p<0.001) and adrenal (51.85%, p<0.001) corticosterone levels in 24 months aged rats without showing any significant alteration of the values (p>0.05) in 18 months aged rats (Table 1), compared to the corresponding observations in 4 months rats treated with vehicle. In young (4 months) rats, carnosine treatment significantly increased (64.72%, p<0.001) plasma corticosterone without significantly affecting the adrenal gland (Table 1) with respect to their corresponding age-matched vehicle-treated rats.
Carnosine and aging-induced enhancement of brain regional Aβ (1-42) steady state level: Carnosine treatment (2.0 µg/Kg/day, i.t.; for 21 consecutive days) significantly lowered the aging (18 and 24 months)-induced enhancement of Aβ (1-42) level in prefrontal cortex (45.70%, p<0.001 and 50.81%, p<0.001 respectively), hippocampus (22.78%, p<0.05 and 62.76%, p<0.001 respectively), hypothalamus (46.31%, p<0.001 in 24 months), and pons-medulla (54.84%, p<0.001 in 24 months) without any significant alteration in hypothalamus and pons-medulla of 18 months and cerebellum of 24 months aged rats with respect to their corresponding vehicle-treated age-matched group of aged rats (Table 2). In 18 months aged rats, Aβ (1-42) level in cerebellum significantly reduced (37.48%, p<0.001) with carnosine. The carnosine-induced attenuation was found to be maximum in hippocampus followed by prefrontal cortex ≥ pons-medulla > hypothalamus in aged (24 months) rats. In young rats (4 months), carnosine did not show any significant effect on brain regional Aβ (1-42) level in comparison to their corresponding results of vehicle-treated young (4 months) rats under the similar conditions (Table 2).
Aging-induced brain regional increase of Aβ plaques and effect of carnosine: Figures 2b-5b and 2c-5c represent a qualitative decrease in aging-induced enhancement of Aβ plaques in all the brain regions after the treatment with carnosine at 2.0 µg/Kg/day (i.t.) for 21 consecutive days in comparison to their corresponding results of age-matched (18 and 24 months) vehicle-treated rats. Figures 2a-5a show that carnosine treatment (2.0 µg/Kg/day, i.t.) for 21 consecutive days quantitatively lowered the aging-induced increase of Aβ plaques in prefrontal cortex (31.58%, p<0.01 and 23.96%, p<0.01 respectively), hippocampal CA1 zone (27.97%, p<0.01 and 24.58%, p<0.01 respectively), hippocampal CA3 zone (33.66%, p<0.01 and 24.24%, p<0.01 respectively), hypothalamus (35.94%, p<0.01 and 26.10%, p<0.01 respectively), and pons-medulla (35.29%, p<0.01 and 24.66%, p<0.01 respectively) of both 18 and 24 months aged rats respectively with respect to their corresponding results of vehicle-treated rats of same age groups.
Aging-induced reduction in rearing count and open field locomotion activity and role of carnosine: The treatment of carnosine (2.0 µg/Kg/day, i.t., for 21 consecutive days) significantly attenuated the aging (24 months)-induced decrease in rearing count and open field locomotion (166.67%, p<0.001 and 113.89%, p<0.001 respectively) with respect to the corresponding results of the vehicle-treated 24 months aged rats (Table 3). However, no significant (p>0.05) effect was observed in 4 months young (9.09% and 14.63% respectively) and 18 months aged (20.00% and 13.95% respectively) rats with carnosine under similar conditions.
Aging-induced stress and various neurological diseases associated with brain regional Aβ deposition and consequent neurodegeneration have been the keen areas of research for the last many decades [8]. Stress from different sources, such as environmental, physical, biochemical, even hormonal during aging makes aging-induced alteration in brain more detrimental and complex and it could be explored with modern analytical perspective. The glucocorticoid (corticosterone in rats) has been found to involve in the hormonal stress [8]. Though, the aging-induced corticosterone status and brain regional pathological deposition of Aβ have long been studied [7,30], the reports on the neurobiological correlation of functional decline of brain regions, associated with Aβ deposition and corticosterone status during normal aging are inadequate. Corticosterone has the potency to induce the Alzheimer’s disease (AD)-like pathology with a reduction in release of different synaptic proteins and their functions in different neural networks and neurodegeneration [31]. Alterations in neuronal network with elevated levels of corticosterone and Aβ deposition have been well reported regarding cognitively impaired aged individuals [32]; whereas, no direct evidence is available regarding locomotor activity in relation to up regulation of stress hormone, because the muscular dystrophy is an another indication of uprise in stress hormone, known as allostatic load [8]. Brain aging, at the level of Aβ-induced neurotoxicity and neurodegeneration has been found to be associated with the reduction in brain regional antioxidant systems [16]. Application of endogenous antioxidant biomolecule, such as carnosine or homocarnosine or anserine, has been found to restore the aging-induced reduction in brain regional antioxidant systems [16]. But the knowledge, regarding their role in aging-induced (a) brain regional Aβ level, and its deposition (qualitatively and quantitatively), (b) endocrine functions (at the level of plasma and adrenal corticosterone status), and (c) behavioural changes (locomotor activity) are not well defined. Carnosine being an antioxidant dipeptide, appears to be a pluripotent biomolecule as it scavanges ROS and RNS [17]. Thus, it is quite reasonable that this unique biomolecule might have some role on neurodegeneration at the level of brain regional Aβ deposition as plaques, corticosterone status in both plasma and adrenal gland, and motor behaviour associated with aging processes.
The corticosterone is known to release from the adrenal cortex into the bloodstream in response to the released corticotrophin-releasing hormone (CRH) and ACTH from the hypothalamus and pituitary respectively [2]. During aging and aging-related neuro diseases, the feedback inhibitory mechanism has been found to be deregulated, and produces more corticosterone in the circulation from the adrenal gland [33]. The aging-induced elevation of plasma corticosterone (Table 1), as observed in the present study seems to act in dual way: (a) the enhancement in Aβ production in the different brain regions (Table 2, Figures 2-5) in one hand, and on the other hand (b) reduction in locomotor activity (Table 3). In addition to these, the aging-related elevation in ROS and RNS makes the scenario more detrimental [34] as presented in Figure 7.
The aging-induced increase in ROS and RNS production from different sources, such as monoamine oxidase-A (MAO-A) enzyme activity [4,34] has been found to damage mitochondrial microenvironment which leads to damage mitochondria [8,34]. Those damaged mitochondria are usually removed and degraded by the autophagic pathway [35]. But the expression of the key regulator of autophagy, baclin1 has been found to reduce during aging which allows to generate more ROS [35]. In conjunction with this increase in ROS and/ or RNS, and brain regional oxidative and nitrosative stress-induced reduction of baclin1 [36] and the aging-induced enhanced plasma corticosterone (Table 1) together might increase the level of APP and β-site APP cleaving enzyme (BACE) to promote the erroneous production of comparatively larger fragments of Aβ (Aβ 1-40 or 1-42) enormously [34] in different brain regions studied (Figure 7). Further, the elevated corticosterone, on other hand has been found to suppress the ability of adult astrocytes and microglia to clear this extracellular Aβ (≤1-40) in the synaptic junctions [34,37] of different brain regions. These larger fragments contain more fibrillogenic property which helps to deposit, and also invites other smaller fragments of Aβ (<1-40) to oligomerize with them [7] to form plaques (Figures 2-5). In this context, it may be mentioned that the brain region-specific knowledge of Aβ deposition during aging has not been studied yet in detail under similar conditions. In the present investigation, in different brain regions this Aβ plaque (both qualitatively and quantitatively) is found to deposit in a graded manner with the maximum Aβ plaque in prefrontal cortex (Figure 2) followed by hippocampal CA1 (Figure 3), hypothalamus (Figure 4), hippocampal CA3 zone (Figure 3) and pons-medulla (Figure 5) whereas, no Aβ deposition has been observed in cerebellum (Figure 6) during aging. These observations may further be strengthened by the aging-induced similar graded increase in steady state level of Aβ (1-42) in the respective brain regions, except cerebellum (Table 2). This aging-induced increase in Aβ deposition may be explained by the fact that the aging-induced impaired insulin activity [38] may also reduce the insulin-sensitive hepatic lipoprotein receptor 1 (LRP1) mediated systemic clearance of Aβ from the respective brain regions [38,39]. This may initiate a positive feedback (Wang et al. 2006) on RAGE (receptor for advanced glycation end product)-dependent influx of free-Aβ from systemic circulation to those brain regions [39] where less protease activity has been found [34,40]. This mechanism might exclude cerebellum (Figure 6) where neprilysin, a protease of Aβ, enzyme activity has been found to be higher in comparison to other brain regions studied [40].
Moreover, the graded significant deposition of Aβ in descending order (prefrontal cortex> hippocampal CA1> hypothalamus> hippocampal CA3> pons-medulla) in those brain regions (Figures 2-6, Tables 1-2) may be explained by (a) the spreading of Aβ in an anterograde direction with a distinct hierarchy (from cerebral cortex or hippocampus to hypothalamus to pons-medulla) which is maintained in the regions that receive neuronal projection (hypothalamus or pons-medulla) from the regions affected (cerebral cortex or hippocampus) by Aβ (1-42) [41], (b) the brain regional different clearance pattern of amyloid beta deposition with the help of reactive astroglia may be due to their distance from plaques [42] and / or (c) less activity of Aβ degradating enzymes [insulin degradating enzyme (IDE) and neprilysin (NEP)], e.g. prefrontal cortex in comparison to cerebellum [43] where no Aβ plaque was observed (Figure 6), despite high level of Aβ (Table 2). As a possible consequence of such Aβ deposition, oxidative stress-induced neurodegeneration and microglial hyperactivity mediated neuroinflammatory cell loss might occur in all the brain regions studied [37,44]. These aging-induced brain regional Aβ deposition (Figures 2-5, Table 2) and consequent neuronal cell loss (unpublished) may further be correlated and strengthen the changes in the functional involvement of aged brain regions including (a) reduction of episodic memory performance related to prefrontal cortex [45], (b) cognitive deficits associated with hippocampal CA1 as well as CA3 pyramidal neuron dysfunction [46], (c) deregulation of the hypothalamo-pituitary-adrenocortical (HPA) axis [47], (d) deterioration of sleep-wake cycle [48] and sympathetic outflow [49] associated with pons-medulla, and (e) deficits in multi-joint motor coordination related to cerebellar dysfunction [14]. In addition to these well established behavioural alterations, the aging-induced increase in cerebrocortical and hippocampal Aβ plaque (Figures 2-3) and Aβ steady state level (Table 2) may be correlated with the increase in plasma corticosterone status (Table 2) and peripheral muscular dysfunction or allostatic load [8] as described earlier. The peripheral muscle of hind and forelimbs might be dystrophic with the aging-induced increase in plasma corticosterone which deflects in the locomotion [50]. The involvement of brain regions (cerebral cortex and hippocampus) with this muscular function may exist, but in this investigation, the role of stress hormone (corticosterone) during aging on the rearing count is an additional support to open field locomotion, together as locomotor activity (Table 3). During aging the allostatic load may be further corroborated and explained with the observation of aging-induced decrease in rearing count and open field locomotion (Table 3) with the increase of plasma corticosterone level (Table 1). Thus, it may be stated that the present observation may seem to relate the fibroblast aging with the brain aging as well.
Carnosine (2.0 μg/Kg/day, i.t., for 21 consecutive days), in the present study, attenuated the aging-induced increase in corticosterone status both in plasma and adrenal gland (Table 1) which seems to reduce the aging-induced stress (ROS and RNS) related microenvironment. This could be due to an alteration of glucocorticoid-mediated aging-induced increase in brain regional MAO-A activity [4]. This aging-induced generation of ROS and RNS may be reduced and the scavenging activity of the antioxidant system may be uplifted due to carnosine supplementation [16,17] as carnosine (β-Ala-L-His) is known to catabolise into alanine and histidine by carnosinase enzyme, and alanine having the higher Km to carnosine synthase to produces carnosine further, whereas histidine produces histamine by histidine decarboxylase [51]. In addition to this, a recent study depicts that carnosine exerts anti-stress activity through this histamine metabolic pathway [51]. The carnosine-induced decrease in aging-related ROS and RNS [17,52] may also restore the aging-induced increase in β- and γ-secretase enzyme activities [17,53] which may lead to down regulate the overproduction of amyloid protein from APP [54]. The attenuation in the secretase (β and γ) activities by carnosine might also dephosphorylate the Aβ [52] which in turn reduces the Aβ plaques both qualitatively and quantitatively (Figures 2-5). This is possible by controlling further accumulation (Table 2) or solubilizing the aggregated/ oligomerized Aβ proteins by uplifting the microglial activity [37,52] and/or the neprilysin activity in the synaptic junctions, though needs further clarification. These explanations may be correlated with the present observation of graded attenuation of aging-induced brain regional Aβ deposition, as observed previously in the order of cerebral cortex> hippocampal CA1> hypothalamus> hippocampal CA3≥ pons-medulla (Figures 2-5, Table 2). In addition, restoration in aging-induced deterioration in brain regional Aβ clearance [39] following the prevention of microglial inflammation [37] by carnosine under similar conditions cannot be ignored. The cytotoxicity due to deposition of Aβ and dysfunctional microglia in synapse has also been found to be reduced with carnosine [17,37]. Since brain region-specific increase in Aβ deposition has been correlated with down regulation of microglial activity [37], the present results (Figures 2-5) indicate the correlation of carnosine mediated activation of microglia and brain-derived neurotrophic factor (BDNF) in those brain regions [37,55] to attenuate the aging-induced increase of Aβ plaque deposition (Figures 2-6). Thus, carnosine under similar conditions does not show any significant effect in either of the young brain regional cellular architecture (Figures 2b-6b) or brain regional steady state level of Aβ (Table 2). Furthermore, the administration of carnosine (2.0 µg/kg/day, for 21 consecutive days) through intrathecally (i.t.) provides carnosine directly into the different regions of the aging brain where the carnosine content is reduced due to increase in carnosinase activity during aging [16]. In this context it may be mentioned that in 24 months carnosine treated aged rats, β-alanine (a hydrolyzed product of carnosine) content increases to a great extent in those brain regions which possibly promote the carnosine synthase activity and recycle itself to synthesize carnosine de novo [13,17] due to higher Km of carnosine synthase activity for its substrate β-alanine [18]. Carnosine treatment (2.0 µg/kg/day, i.t.; for 21 consecutive days) may upgrade the endogenous carnosine level [13] with its (carnosine) potentiation in fibrinolytic activity on Aβ plaque (Figures 2-5) as a neuroprotectant biomolecule [17]. In addition, the reduction in aging-induced peripheral neuromuscular activity has been found to attenuate with the reduction of aging-induced increase in plasma corticosterone in both the rearing count and open filed locomotion (Table 3) with carnosine and hence might be attenuated the fibroblast aging [56] / allostatic load (Table 2) in relation to the respective brain regions during aging.
In conclusion, (a) aging-induced brain regional neurodegeneration in terms of their Aβ (1-42) level (Table 2) and its depositions (Figures 2-5), and neuronal cell loss (unpublished) may be correlated with the region-specific functional deterioration of locomotor activity in relation to the up regulation of plasma corticosterone status and (b) carnosine (2.0 μg/Kg/day, i.t.; for 21 consecutive days) attenuates these aging-induced phenomena to an extent that is comparable with young (4 months) rats.
Carnosine attenuated aging-induced increase in plasma & adrenal corticosterone level.
Graded accumulation of Aβ plaque in different brain regions with aging.
Qualitative & quantitative attenuation of aging-induced brain regional Aβ with carnosine.
Aging-induced deterioration of behavioral activity attenuated with carnosine.
We are thankful to Prof. S. Sengupta, Department of Statistics, University of Calcutta for helping the statistical analysis of the data; Dr. Samir K. Ghosh for his technical help in IHC staining. The present research work was financially supported by the National Post-Doctoral Fellowship (NPDF) of Science and Engineering Research Board (SERB), Govt. of India; the University Grants Commission (UGC), New Delhi, India; Indian Council of Medical Research (ICMR), New Delhi, India.
The authors declare no conflict of interest.
S.B., B.M. and M.K.P. made substantial contributions to the study concept and design, acquisition of the data, and analysis and interpretation of the data. S.B. participated in drafting the Article, B.M. and M.K.P. were involved in critically revising it for important intellectual content. All authors gave final approval of the version before submission.
- Hayflick L (2000) The future of ageing. Nature 408: 267-269. [Crossref]
- Ulrich-Lai YM1, Herman JP (2009) Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci 10: 397-409. [Crossref]
- Bush IE, Sandberg AA (1953) Adrenocortical hormones in human plasma. J Biol Chem 205: 783-793. [Crossref]
- Banerjee S, Poddar MK (2015) Carnosine: effect on aging-induced increase in brain regional monoamine oxidase-A activity. Neurosci Res 92: 62-70. [Crossref]
- Swaab DF, Bao AM, Lucassen PJ (2005) The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev 4: 141-194. [Crossref]
- de Kloet ER, Vreugdenhil E, Oitzl MS, Joels M (1998) Brain corticosteroid receptor balance in health and disease. Endocr Rev 19: 269-301.
- Dong H, Yuedea CM, Yooa HS, Martina MV, Deala C, Macea AG, Csernansky JG (2008) Corticosterone and related receptor expression are associated with increased ß-amyloid plaques in isolated Tg2576 mice. Neurosci 155: 154-163.
- Garrido P (2011) Aging and stress: past hypotheses, present approaches and perspectives. Aging Dis 2: 80-99. [Crossref]
- Scahill RI, Frost C, Jenkins R, Whitwell JL, Rossor MN, Fox NC (2003) A longitudinal study of brain volume changes in normal aging using serial registered magnetic resonance imaging. Arch Neurol 60: 989-994.
- Golde TE, Miller VM (2009) Proteinopathy-induced neuronal senescence: a hypothesis for brain failure in Alzheimer's and other neurodegenerative diseases. Alz Res Ther 1: 5.
- Selkoe DJ (1998) The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol 8: 447-453. [Crossref]
- Fukumoto H, Asami-Odaka A, Suzuki N, Shimada H, Ihara Y, Iwatsubo T (1996) Amyloid ß Protein Deposition in Normal Aging Has the Same Characteristics as That in Alzheimer's Disease: Predominance of Aß42 (43) and Association of Aß40 with Cored Plaques. Am J Pathol 14: 259-265.
- Banerjee S, Poddar MK (2016) Aging-induced changes in brain regional receptor binding: effect of Carnosine. Neurosci 319: 79-91.
- Raz N, Lindenberger U, Rodrigue KM, Kennedy KM, Head D et al. (2005) Regional brain changes in aging healthy adults: general trends, individual differences and modifiers. Cereb Cortex 15: 1676-1689.
- Sonani RR, Rastogi RP, Madamwar D (2015) Antioxidant potential of phycobiliproteins: Role in anti-aging research. Biochem. Analytical Biochem 4: 172.
- Bellia F, Vecchio G, Cuzzocrea S, Calabrese V, Rizzarelli E (2011) Neuroprotective features of carnosine in oxidative driven diseases. Mol Aspects Med 32: 258-266.
- Boldyrev AA, Aldini G, Derave W (2013) Physiology and pathophysiology of carnosine. Physiol Rev 93: 1803-1845. [Crossref]
- Sale C, Saunders B, Harris RC (2010) Effect of beta-alanine supplementation on muscle carnosine concentrations and exercise performance. Amino Acids 39: 321-333.
- Drozak J, Veiga-da-Cunha M, Vertommen D, Stroobant V, Van SE (2010) Molecular identification of carnosine synthase as ATP-grasp domain containing protein 1 (ATPGD1). J Biol Chem 285: 9346-9356.
- Margolis FL (1974) Carnosine in the primary olfactory pathway. Science 184: 909-911. [Crossref]
- Banerjee S, Ghosh TK, Poddar MK (2015) Carnosine reverses the aging-induced down regulation of brain regional serotonergic system. Mech Ageing Dev 152: 5-14. [Crossref]
- Bae OK, Seung-Hoon BK, Lee KY, Dorrance A, Rumbeiha W, Fitzgerald SD et al. (2013) Safety and efficacy evaluation of carnosine, an endogenous neuroprotective agent for ischemic stroke. Stroke 44: 205-212.
- Banerjee S, Poddar MK (2013) Platelet monoamine oxidase-A activity and aging: effect of carnosine. J Physiol Sci 63: 279-285. [Crossref]
- Bandyopadhyay BC, Poddar MK (1998) Dietary protein-induced change in mammalian corticosterone status (index of immune response) during aging. Mech Ageing Dev 103: 57-68. [Crossref]
- Poddar MK, Dewey WL (1980) Effect of cannabinoids on catecholamine uptake and release in hypothalamus and striatal synaptosomes. J Pharmacol Exp Ther 214: 63-67.
- Aho L, Pikkarainen M, Hiltunen M, Leinonen V, Alafuzoff I (2010) Immunohistochemical visualization of amyloid-ß protein precursor and amyloid-ß in extra- and intracellular compartments in the human brain. J Alz Dis 20: 1015-1028.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265-275. [Crossref]
- Dalal A, Poddar MK (2009) Short-term erythrosine B-induced inhibition of the brain regional serotonergic activity suppresses motor activity (exploratory behaviour) of young adult mammals. Pharmacol Biochem Behav 92: 574-582.
- Turner KM, Burne THJ (2014) Comprehensive behavioural analysis of long evans and sprague-dawley rats reveals differential effects of housing conditions on tests relevant to neuropsychiatric disorders. PLoS ONE 9: e93411.
- Csernansky JG, Dong H, Fagan AM, Wang L, Xiong C, et al. (2006) Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am J Psychiatry 163: 2164-2169. [Crossref]
- Wuwongse S, Cheng SS-Y, Wong GT-H, Hung CH-L, Zhang NQ, Ho Y-S, et al. (2013) Effects of corticosterone and amyloid-beta on proteins essential for synaptic function: Implications for depression and Alzheimer's disease. Biochimica et Biophysica Acta 1832: 2245-2256.
- Sperling RA, Laviolette PS, O'Keefe K, O'Brien J, Rentz DM, Pihlajamaki M, et al. (2009) Amyloid Deposition Is Associated with Impaired Default Network Function in Older Persons without Dementia. Neuron 63: 178-188.
- Lo MJ, Kau MM, Cho WL, Wang PS (2000) Aging effects on the secretion of corticosterone in male rats. J Investig Med 48: 335-342. [Crossref]
- Wang Y, Li M, Tang J, Song M, Xu X, Xiong J, et al. (2011) Glucocorticoids facilitate astrocytic amyloid-ß peptide deposition by increasing the expression of APP and BACE1 and decreasing the expression of amyloid-ß degrading proteases. Endocrinol 152: 2704-2715.
- Brunk UT, Terman A (2002) The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem 269: 1996-2002. [Crossref]
- Zhang Z, Mingke S, Xia L, Seong SK, Duc MD, Nicholas TS, et al. (2015) Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat Commun 6: 8762.
- Caruso G, Fresta CG, Musso N, Giambirtone M, Grasso M, Spampinato SF, et al. (2019) Carnosine Prevents Aß-Induced Oxidative Stress and Inflammation in Microglial Cells: A Key Role of TGF-ß1. Cells 8: 64.
- Mazumdar R, Xiaohui M, Gil A, Patricia V, Xiaoman Y, Nir B, et al. (2004) Decrease in Glucose-Stimulated Insulin Secretion with Aging Is Independent of Insulin Action. Diabetes 53: 411-446.
- Deane RR, Bell D, Sagare A, Zlokovic BV (2009) Clearance of amyloid-ß peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol Disorder-Drug Targets 8: 16-30.
- Yasojima K, McGeer EG, McGeer PL (2001) Relationship between beta amyloid peptide generating molecules and neprilysin in Alzheimer disease and normal brain. Brain Res 919: 115-121.
- Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58: 1791-1800. [Crossref]
- Olabarria M, Noristani HN, Verkhratsky A, Rodríguez JJ (2010) Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58: 831-838.
- Jing D, Bing S, Kui C, Lang Z, Shubo L, Qingquan G et al. (2009) Metabolites of cerebellar neurons and hippocampal neurons play opposite roles in pathogenesis of Alzheimer's disease. PLoS ONE 4: e5530.
- Hong X, Steven H, Jun J, Maria S, Jonathan K, Brian JB, et al. (2013) Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc Nat Acad Sci (USA) 110: 7904-7909.
- Simone R, Carlo M, Patrizio P, Claudio B, Paolo MR, Stefano FC, et al. (2004) Age-Related Functional Changes of Prefrontal Cortex in Long-Term Memory: A Repetitive Transcranial Magnetic Stimulation Study. J Neurosci 24: 7939-7944.
- Simkin D, Hattori S, Ybarra N, Musial TF, Buss EW, Richter H, et al. (2015) Aging-Related Hyperexcitability in CA3 Pyramidal Neurons is Mediated by Enhanced A-Type K+ Channel Function and Expression. J Neurosci 35: 13206-13218.
- Brureau A, Zussy C, Delair B, Ogier C, Ixart G, Maurice T, Givalois L (2013) Deregulation of hypothalamic-pituitary-adrenal axis functions in an Alzheimer’s disease rat model. Neurobiol Aging 34: 1426-1439.
- Hut RA, Van der Zee EA (2011) The cholinergic system circadian rhythmicity and time memory. Behav Brain Res 221: 466-480.
- Samuels ER, Szabadi E (2008) Functional neuroanatomy of the noradrenergic locus ceruleus: its roles in the regulation of arousal and autonomic function partII: physiological andpharmacological manipulations and pathological alterations of locuscoeruleus activity in humans. Curr Neuropharmacol 6: 254-285.
- Elias PK, Redgate E (1975) Effects of immobilization stress on open field behavior and plasma corticosterone levels of aging C57BL/6J mice. Experimental Aging Res 1: 127-135.
- Li YF, He RR, Tsoi B, Li XD, Li WX, et al. (2012) Anti-stress effects of carnosine on restraint-evoked immunocompromise in mice through spleen lymphocyte number maintenance. PLoS One 7: e33190. [Crossref]
- Wu JW, Liu K-N, How S-C, Chen W-A, Lai C-M, Liu H-W, et al. (2013) Carnosine’s Effect on Amyloid Fibril Formation and Induced Cytotoxicity of Lysozyme. PLoS ONE 8: e81982.
- Zimmermann M, Gardoni F, Di Luca M (2005) Molecular rationale for the pharmacological treatment of Alzheimer's disease. Drugs & Aging 1: 27-37.
- Hipkiss AR (2007) Could carnosine or related structures suppress Alzheimer's disease? J Alzheimers Dis 11: 229-240. [Crossref]
- Murakami T, Furuse M (2010) The impact of taurine- and beta-alanine supplemented diets on behavioral and neurochemical parameters in mice: antidepressant versus anxiolytic-like effects. Amino Acids 39: 427-434.
- McFarland GA, Holliday R (1999) Further evidence for the rejuvenating effects of the dipeptide L-carnosine on cultured human diploid fibroblasts. Exp Gerontol 34: 35-45. [Crossref]