Breast cancer, a complex and heterogenous disease is a common malignancy in women. The disease is linked to deregulation of cell cycle controls apart from other reasons, like genetic predisposition, and or environmental causes. Identification of the key molecules involved in cell cycle deregulation can help to design targeted therapies using small molecule inhibitors. The targeted therapeutic agents can prove to be not only safe but also effective in cancer treatment and management.
breast cancer, cell cycle regulation, cell cycle check points, spindle assembly, neosis, targeted therapy
Breast cancer, a complex and heterogenous disease of different types often poses difficult situations for treatment [1,2]. Though there is remarkable improvement in diagnostic tools and therapeutic options, cure rates in breast cancer remains poor. The GLOBOCAN-2018 report, reports 162,000 breast cancer cases (27.7% of all new cancers), a 5-year prevalence of 405,000 breast cancer cases, and 87,000 deaths were observed in the Indian population [3]. Biomarkers, oncogenes and tumour suppressor genes can help in the therapeutic decisions. The knowledge of cell cycle and cell cycle components can help in development of reliable biomarkers, and accurate measurement of cell cycle component expression and their correlation with clinical symptoms and prognosis can give insight for the future of both breast cancer management and anti-cancer therapeutics [4]. Targeted therapy or precision drug therapy involves use of drugs, or knowledge of cell cycle components that influence not only the cancer cell growth, but also survival and proliferation. This review gives an insight into the cell cycle and its components and targeted therapy as an option for breast cancer remedies.
The cell cycle is a fundamental and inevitable pathway of events that plays role in the cellular physiology, development and diseases. Regulation of this cyclical process at different stages like in mitosis involves number of proteins which eventually leads to the formation of daughter cells. The regulatory process also involves cyclin dependent kinases (CDKs) and cyclins which control passage of the cycle through different stages namely, G1, G0, S, G2 and M phases. Kinases inturn can be regulated by CDK inhibitors (CDKIs) [5]. The cyclical period of division of somatic cells can be grouped under two heads: (i) Interphase and (ii) Mitosis (M phase). On the other side, germ cells undergo broadly two stages, namely nuclear and cell division stages respectively named as Meiosis I and Meiosis II. Interphase, a preparatory period of cell division can be further subdivided to G1, (G0 in dormant cells) S and G2 phase [6]. In turn mitosis, the period of cell division consists of prophase, metaphase, anaphase and telophase. The CDKs and cyclins play role at every stage of the cycle to check for errors and necessary corrections. However, defects can arise in the pathway molecules due to different reasons, like gene mutations, and can result in development of diseases like cancer [7].
Cell growth
The process of cell division results in increase in cell number which is a part of cellular growth [8]. Cell growth can happen either due to cellular proliferation resulting in increase in cell numbers, or by increase in cell volume occurring in absence of cell division. Cell growth can be influenced by a number of factors, like leukocyte infiltration [9,10]. However, growth and cell cycle can be independently regulated and cell cycle does not drive cell growth. This might be due to the fact that cell can often divide without growing, like disproportionate division of cytoplasm to small cells in egg cells [11-13]. Further to control cell size, controls are imposed by developmental programs upon cell growth and cell proliferation. This could be regulated by the divergence of regulatory pathways governing growth and proliferation [14].
The regulation of cell cycle done by cyclins and CDKs are important in both self –renewing and transit amplifying divisions [15]. However, as metazoan cells progress through development their cells progress through various types of cell cycle, like embryonic cell cycle, meiotic and mitotic cell cycle, endoreduplication cell cycle [16]. The factors required in each cycle progression have differential requirements of critical regulators as per the respective cycles. In turn, check point controls can arrest the cell cycle progression. For instance, the cell cycle might be arrested in response to DNA damage and cell death might be initiated. The DNA damage checkpoint, replication checkpoints, spindle-assembly check point might impose brakes on cell cycle and initiate for correction of the stages before allowing the cycle to proceed further [17,18]. There might be influence of the environmental conditions and mutations to interrupt the reproducible pattern of cell division as observed in C. elegans. Though cell-cycle regulation interconnects with developmental stages, many more things are to be elucidated. Proliferations at germline level along with the control mechanisms involve some of the best characterized connections between developmental signals and cell-division control (Figure 1) [19,20]. However, it needs to be looked more at how developmental programs control the cell cycle and whether systemic signals are involved [21].

Figure 1. Schematic illustration of cell proliferation mechanism
Cancer cells undergo cell division by a unique way called as neosis, through which they could escape senescence and enter to transformation and progression of tumours. However, cells can have induction of senescent phenotype by either, telomere attrition-induced senescence at end of cellular mitotic life span (MLS), or by replication history-independent, accelerated senescence by inadvertent activation of oncogenes or by exposure of cells to genotoxins [21]. Senescent cells with mutations or epimutations in tumour suppressor genes would escape cell death by mitotic catastrophe and become polyploid cells, which before their death gives rise to many cells with viable genomes by nuclear budding and asymmetric cytokinesis, or development of genetic instability [22,23]. The mode of cell division occurred here is called as ‘neosis’ and the immediate neotic progeny are called as ‘Raju cells’ and display stem cell properties. However, the offsprings show a limited MLS and enter senescence. Raju cells undergo mitotic division and can subsequently show resistance to genotoxins leading to generation of drug resistant tumour growth (Figure 2) [24]. In brief, neosis can extend MLS of cells which are not favouring mitosis, preceds tumourigenesis, occur multiple times during tumour progression, generating Raju cells and conferring tumour cell heterogeneity that would be further subjected to natural selection during tumour progression and can involve epigenetic modulations [25].
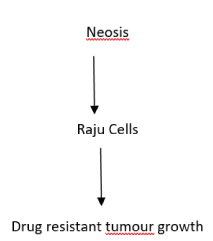
Figure 2. Illustration of Neosis
Triple negative breast cancer
Hormonal therapy could be an option for cancer treatment but in breast cancer there is an added problem and it is about the absence of hormone receptors (estrogen, progesterone, and HER2 protein) leading to a state being developed called as triple negative breast cancer [26]. This state might be an ideal predisposition for chemotherapy treatment but preferably in the early stages.
Cell cycle regulation
Eukaryotic cells undergo division. The regulatory process is understood from the experimental on different organisms. The cell cycle consists of divisive phases/events of nucleus and the cytoplasm. The progression through the phases is controlled by the cyclin dependent kinases (CDKs). The entry of oocytes to meiosis, or oocyte maturation is regulated by the cytoplasmic factor, also called as the maturation promoting factor (MPF) which is also present in somatic cells [27]. Cell cycle regulation process is influenced drastically by the protein phosphorylation mediated by cdc2 (cdc stands for cell division cycle mutant) encoded protein kinases. The other group of proteins which will be influencing the process are called as cyclins which display a periodic pattern of accumulation and degradation in the cells. The MPF is regulated by phosphorylation and dephosphorylation of Cdc2, and in turn the catalytic activity of Cdc2 protein kinase is regulated by cyclin B. The Cdc2 kinase activity is initiated by Cdc2 phosphorylation at threonine-161 and tyrosine 15 positions and threonine 14 in vertebrates. Dephosphorylation of threonine-14 and tyrosine-15 by a protein phosphatase, Cdc25 leads to the activation of Cdc2/cyclin B complex and transition from G2 to M phase of the cell cycle. Furthermore, Cdc2 activity by ubiquitylation leads to the degradation of cyclin B, inturn leading to inactivation of Cdc2, helping the cells to exit mitosis, undergo cytokinesis and return back to interphase [27,28]. Although control of the G2/M transition is implicated in events in cancer resulting in chromosomal aberrations, important cell-cycle events in G1/S transition may be specifically altered in breast cancer, including the actions of the oncogenes/ tumor suppressors cyclin E, cyclin D1, and p27 [29]. The Cdc2 related protein kinases are called as cyclin dependant kinases (Cdks). Progression from G1 to S is regulated by Cdk2 and Cdk4, or Cdk6 with cyclins D and E, whereas progression through the restriction point in G1 is mediated by the complex of Cdk 4 and 6 with Cyclin D. The C phase is mediated by activation of Cdk2/cyclin A complex. Additionally, Cdk/cyclin complex, like Cdk7/cyclin H plays role not only in cell cycle regulation, but also in transcription, because of its association with transcription factor TFIIH [30].
The steroid hormone estrogen is hypothesized to induce cell-cycle progression either through c-Myc, or through cyclin D1 [31]. Sequestration of p21 and p27 by cyclin D1 and Cdk4 leads to the activation of cyclin E/Cdk2 by estrogen and downregulated p21 levels in presence of c-Myc [32,33]. Both c-Myc and cyclin D1 can be overexpressed by progestin [31]. Prolactin and its receptor also acts as an important physiological regulator of mammary epithelial cell activity, and prolactin treatment of cultured breast cancer cells lead to the induction of cyclin D1 and decreased expression of p21, but not p27 [34].
Mammary epithelial cells can also have inhibition of G1/S activity due to cytokine TGF-β activity by induction of CDK inhibitors and modulation of CDK activity [31]. Upregulation of cyclin D is linked not only to the resistance development against endocrine therapy in breast cancer, but also to the overexpression (in 20% of breast cancer incidences), and copy number amplification (in 50% of breast cancer incidences) apart from more expression of cyclin D1 in 67.5% of invasive ductal carcinoma incidences [35-38]. Genetic alterations in the cyclin D1 gene and mRNA expression was seen in the ER-negative MDA-MB-453 cell line, which can lead to malignancy [39]. In another study, ER-positive breast cancer patients were reported to have high cyclin D1 expression, with more risk of relapse, metastasis, and early death [40,41]. Cyclin D1 was reported not only to be higher in proliferative disease, but also in ductal carcinoma in situ, and also overexpessed in invasive lobular carcinomas [42,43]. Cyclin D3 was also seen to be associated with breast cancer samples [4].
Expression levels of cyclin A with/without E2-promoter binding factor 1 (E2F1) associated with Ki-67 can be used as a marker in breast cancer disease analysis. Even, overexpressed cyclin A can be correlated with earlier relapse, higher risk, and shorter overall survival rate in breast cancer patients [44-46]. Gene amplification of cyclin E has been seen also in breast cancer tissues with more risk to the breast cancer patients [47,4], and cyclin E may be responsible for ER-independent tumour growth [48,49]. Furthermore, cyclin B was found to be associated with lymphatic invasion and poor survival under breast cancer disease condition, and cyclin B1 is considered to be a strong prognostic factor in breast cancer [4,50]. Of the tumour suppressors, aberrant p16 gene expression, closely associated with a high proliferative index is one of the most common abnormality in breast cancer [51,52]. However, low p16 gene expression is also seen in TNBC carcinoma [53]. Expression of p21 can act as a tumour suppressor, and expression of p21WAF1/CIP1 can be used as a key prognostic marker for breast cancer [54,55]. Again, downregulation of p21WAF1/CIP1 promoted EMT, enhanced the cell viability and migration potential in response to long non-coding RNA plasmacytoma variant translocation 1 (PVT1) in distinct MDA-MB-231, MDA-BA-468 breast cancer cell lines, and also could inhibit apoptosis [4,56]. Upregulation of p21WAF1/CIP1 was seen to be associated with invasion and poor response to tamoxifen tretatment in MCF-7 cells [57]. Low levels of p27 protein also acts as a marker in lymph-node negative breast cancer patients [58]. Elevated levels of p27 in human breast cancer cells inversely correlated with the degree of malignancy in human breast, and significantly associated with ER-positive state [59,60]. Reduced expression of p27 alongwith elevated expression of HER2 could be significantly for HER2-positive/neu antibody therapy, and can also be correlated with a high histologic grade, an advanced TNM stage (tumor size, lymph node status, metastatic status), and negative hormone receptor status [61-63]. The HER 2-positive and TNBC subtypes were also reported to have p53 overexpression [64], suggesting its role as marker in poor prognosis and a compromised immune response in more aggressive breast cancer types [65]. Furthermore, p53 overexpression was found to be inversely correlated with ER/PR expression and positively correlated with HER2-positive overexpression in high-grade tumours with nodal metastasis [66].
Spindle assembly checkpoints
Genetically identical daughter cells are being produced through cell division. The process needs genome replication and chromosome segregation. The spindle assembly checkpoint (SAC) or ‘mitotic checkpoint’ acts to maintain stability by preventing anaphase to proceed till all the kinetochores are stably attached to the spindle fiber microtubules . With stable attachment to the spindle, checkpoint is inactivated and cell cycle progresses [67]. Cyclin B1 and Cdc20 synthesis are essential for maintenance of the mitotic state. The mitotic checkpoint complex (MCC) is composed of the SAC proteins BubR1, Bub3 and Mad2 associated with the Cdc20. This complex acts as an activator of the E3 ubiquitin ligase anaphase promoting complex or cyclosome (APC/CCdc20) [68]. With alignment of all chromosomes at the metaphase plate, SAC silencing is initiated wherein the existing MCC becomes abolished [69,70]. This leads to Cdc20 to be freed and activating APC/CCdc20 leading further to degradation of securing and cyclin B [68]. The cyclin B1 breakdown initiates mitotic exit [68,71,72]. Mitotic slippage or mitotic exit leads to cell death occurring only after one cell cycle following mitotic exit and increasing progressively during subsequent cell cycles [73].
Aneuploidy, common phenomenon in 80% of the breast cancers is linked to the damages in SAC and occurs due to the changes in SAC protein levels and loss of checkpoint functions [70,74,75] and SAC inhibition in aneuploid cells can lead to the generation of mitotic defects [76]. Thus, targeting deregulated SAC can be part of breast cancer therapeutics [68]. Thus, SAC deregulation can lead to chromosomal instability (CIN), and SAC genes are frequently altered at the transcriptional level, whereas mutations at the DNA level are rare [70]. The SAC control also exists in meiosis, and SAC kinases important for mitotic SAC control does additional role in meiosis [77]. The triple negative breast cancer tumours (TNBC) show a high level of CIN, overexpressing mostly the spindle assembly checkpoint kinase TTK, suggesting also TTK inhibition as a therapeutic target [78].
Targeted therapies
Targeted therapy in cancer involves use of drugs to target specific genes and proteins involved in growth and survival of cancer cells. This can help cancer cells to grow in an environment or can target cells involved in cancer growth. The process involves a look out for the involved genetic changes in tumour growth, and a potential target can be a protein with or without a mutation present in cancer cells, but not in the normal healthy cell [79,80]. The therapeutic procedure can lead to destruction of cancer cells, as well as prevent growth and proliferation of cancer cells. Breast cancer can have HER2/neu protein as a target for therapeutic procedures [2,81], apart from inhibitors for CDK4/6, mTOR, and PI3K pathway. PARP inhibitors can be used for situations with BRCA1 gene mutations [82]. Chemotherapy can be combined with targeted therapy for HER2 positive breast cancer patients [83]. Drugs, like olaparib and talazoparib can be used for chemotherapeutic treated patients with HER2-negative breast cancer with BRCA gene mutation. Monoclonal antibodies can be used for patients with TNBC. Hormone therapy can be used for patients with hormone receptor as positive [84].
Targeted therapy differs from the standard chemotherapy in many ways, like wherein the standard chemotherapeutic agents can be cytotoxic, targeted therapeutics can be cytostatic. Targeted therapy acts as precision medicine as these act on the specific molecular targets in cancer, but chemotherapeutic agents acts on all rapidly dividing normal and cancerous cells [85]. In a summary, targeted therapeutics are less likely to harm normal healthy cells than the chemotherapeutic agents [https://www.breastcancer.org/treatment/targeted_therapies]. Most of the targeted therapeutics involve use of small molecules drugs or monoclonal antibodies [86] or use tumour agnostic treatments [87].
Breast cancer, a common cancer found in women mostly, as well as in men [88,89] is a complex disease with many subtypes [1,2]. Though there are targeted therapies for the specific pathophysiology of breast cancers, further research needs to look at the evasion of therapeutic resistance mechanisms, suggesting the need to target the cell cycle molecules.
Most of the therapeutic approaches in cancer target molecules to damage the cell division process. The process leads to activation of cell cycle checkpoints, target repair mechanism, or cell death phase [90]. Tumour metastasis is closely related to the cell cycle [91]. Activities in cell cycle can result in cytoskeletal changes [92]. Epithelial mesenchymal transition is also related to the tumour metastasis [93-96]. Efficacy of chemotherapeutic drugs can be improved alongwith the regulation of cell cycle [91]. Association of p27 with CDK2-cyclin A leads to blocking of putative protein substrate docking region on cyclin A. Thus, structured and designed small molecules can block CDK activity and arrest cell cycle of the cancer cells [97]. The cell death kinase inhibitors with adenovirus can serve not only as vehicle for delivery, but also can lead to CDK inhibition. Examples include effective introduction of p16 INK4a with pRb and adenovirus with p21cip1/waf1 or p27kip1 in cancer cell lines, as well as use of small peptides to mimic endogenous CKIs [98-101]. Reports suggest use of chemical CDK inhibitors to block CDK activity [102]. The regulatory pathways can be altered by using antisense technologies [103,104]. Antibody inhibition of CDC25A in HeLa cells resulted in the formation of mitotic-like phenotype and cell death [105], whereas inhibition of CDC25B lead to S-phase delay and antiproliferative effects [106], resulting in alteration of proteasomal pathways, but can result in complications. Though more information is required, the basic regulatory mechanisms of cell cycle throw up light for discovery of molecules that can target aberrant proliferation [102]. MicroRNAs play important role in cancer [107], and miRNA like MRX34, a miR-34 mimic can be used as the other alternative therapeutic strategy. Though combination therapy with several selective inhibitors can show clinical activity, these can lead to intolerable adverse effects. Thus, there is a requirement to look out for the alternative strategies, like use of predictive biomarker, or biomolecules, including CKIs (Figure 3) [90,108,109].
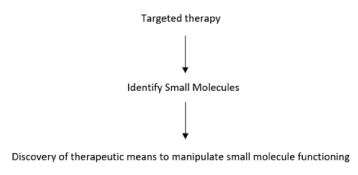
Figure 3. Schematic illustration of targeted therapy concept
Metastatic cancer is the complexity in cancer and can involve SAC deregulation mechanism [68]. Treatment options for metastatic cancer vary on different parameters [87] and SAC can be also hypothesized for therapeutic target [110]. However, a patient needs to be tested for a tumour with drug target by ways like biomarker testing [86], and targeted therapy needs the identification of targets that play important role in cancer cell growth and survival. The lead compounds (compounds that affect the target) are chemically modified the produce closely related versions of the lead compound, which are then tested to determine the effective and fewest effects on nontarget molecules [85].
Targeted cancer therapy involves use of drugs or any other substance that can block the growth and spread of cancer by interfering with molecular targets [85,111]. However targeted therapy needs to work in where a tumour has a target for the drug and can fail if cells become resistant to the targeted therapy [79]. Overcoming resistance development in breast cancer targeted therapies is one of the challenging area in research with combination of different molecules targeting signalling pathways [1]. Systemic therapy can also be an option [87]. Though there are proven drugs, new drugs has been developed for combined treatment strategies against HER2. Herein is the need to research on key genes effecting proliferation and metastasis of breast cancer cells [2]. With increase in incidences of cancer and especially breast cancer [111] targeted therapy can work in circumstances wherein chemotherapy will not work [82].
The author declares no conflict of interest.
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sector.
- Masoud V, Pagès G (2017) Targeted therapies in breast cancer: New challenges to fight against resistance. World J Clin Oncol 8: 120-134.
- Wang J, Xu B (2019) Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduction and Targeted Therapy 4: 34.
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2018) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J Clin 68: 394-424.
- Kashyap D, Garg VK, Sandberg EN, Goel N, Bishayee A (2021) Oncogenic and Tumor suppressive components of the cell cycle in breast cancer progression and prognosis. Pharmaceutics 13: 569.
- Mills CC, Kolb EA, Sampson VB (2018) Development of chemotherapy with cell-cycle inhibitors for adult and pediatric cancer therapy. Cancer Res 78: 2018.
- Schafer KA (1998) The cell cycle: A review. Wyeth-Ayerst Research, Chazy, NY 35: 461-478.
- Shah MA, Schwartz GK (2001) Cell Cycle-mediated drug resistance: an emerging concept in cancer therapy. Clinical Cancer Res 7: 2168-2181.
- Alberts B, Johnson A, Lewis J (2002) Extracellular control of cell division, Cell growth, and apoptosis, in molecular biology of the cell. 4th edition. New York: Garland Science.
- Guo S, Dipietro LA (2010) Factors affecting wound healing. J Dent Res 89: 219-229.
- Schultz GS, Chin GA, Moldawer L (2011) Principles of Wound Healing, in: Fitridge R, Thompson M, [Eds.], Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists [Internet]. Adelaide (AU): University of Adelaide Press. 23.
- Gilbert SF (2000) Developmental Biology. 6th edition. Sunderland (MA): Sinauer Associates. An Introduction to Early Developmental Processes.
- O’Farrell PH (2015) Growing an embryo from a single cell: a hurdle in animal life. Cold Spring Harb Perspect Biol 7: a019042.
- Su TT, O’Farrell PH (1998) Size control: Cell proliferation does not equal growth. Curr Biol 24: 8.
- Zhu J, Thompson CB (2019) Metabolic regulation of cell growth and proliferation. Nature Rev Molecular Cell Biol 20: 436-450.
- Valli H, Phillips BT, Orwig KE, Gassei K, Nagano MC (2015) Chapter 15 - Spermatogonial stem cells and spermatogenesis. In Plant TM, Zeleznik AJ (Eds.), Knobil and Neill's Physiology of Reproduction (Fourth Edition), Academic Press. pp. 595-635.
- van den Heuvel S (2005) Cell-cycle regulation, WormBook, In The C. elegans Research Community (Ed.), WormBook.
- Lawrence KS, Chau T, Engebrecht J (2015) DNA damage response and spindle assembly checkpoint function throughout the cell cycle to ensure genomic integrity. PLoS Genet 11: e1005150.
- Chow JPH, Siu WY, Fung TK, Chan WM, Lau A, et al. (2003) DNA damage during the spindle-assembly checkpoint degrades CDC25A, inhibits cyclin-CDC2 complexes, and reverses cells to interphase. Mol Biol Of the Cell 14: 3989-4002.
- Kimble J, Crittenden SL (2005) Germline proliferation and its control. In WormBook: The Online Review of C. elegans Biology [Internet]. Pasadena (CA): WormBook.
- Hinnat TD, Merkle JA, Ables ET (2020) Coordinating proliferation, polarity, and cell fate in the drosophila female germline. Front Cell Dev Biol 8: 19.
- Park M, Krause MW (1999) Regulation of postembryonic G(1) cell cycle progression in Caenorhabditis elegans by a cyclin D/CDK-like complex. Development 126: 4849-4860.
- Rajaraman R, Guernsey DL, Rajaraman MM, Rajaraman SR (2006) Stem cells, senescence, neosis and self-renewal in cancer. Cancer Cell Intl 6: 25.
- Sundaram M, Guernsey DL, Rajaraman MM, Rajaraman R (2004) Neosis: a novel type of cell division in cancer. Cancer Biology and Therapy 3: 207-218.
- Rajaraman R, Rajaraman MM, Rajaraman SR, Guernsey DL (2005) Neosis--a paradigm of self-renewal in cancer. Cell Biol Intl 29: 1084-1097.
- Rajaraman R, Guernsey DL, Rajaraman MM, Rajaraman SR (2017) Neosis - A parasexual somatic reduction division in cancer. Intl J Human Genetics 7: 29-48.
- Chavez KJ, Garimella SV, Lipkowitz S (2010) Triple negative breast cancer cell lines: one tool in the search for better treatment of triple negative breast cancer. NIH Public Access 32: 35-48.
- Cooper GM (2000) The Cell: A molecular approach. 2nd edition. Sunderland (MA): Sinauer associates. Regulators of cell cycle progression.
- Notes from lecture 8 of Harvard Extension’s Cell Biology course 2021 (accessed 3 October 2021).
- Funk JO (2005) Cell Cycle Checkpoint Genes and Cancer. Encyclopedia of Life Sciences.
- Caldon CE, Daly RJ, Sutherland RL, Musgrove EA (2006) Cell cycle control in breast cancer cells. Journal of Cellular Biochemistry 97: 261-274.
- Foster JS, Henley DC, Ahamed S, Wimalasena J (2001) Estrogens and cell-cycle regulation in breast cancer. Trends Endocrinol Metab 12: 320-327.
- Schroeder MD, Symowicz J, Schuler LA (2002) PRL modulates cell cycle regulators in mammary tumor epithelial cells. Mol Endocrinol 16: 45-57.
- Barnes DM, Gillett CE (1998) Cyclin D1 in breast cancer. Breast Cancer Res Treat 52: 1-15.
- Velasco-Velázquez MA, Li Z, Casimiro M, Loro E, Homsi N, et al. (2011) Examining the role of cyclin D1 in breast cancer. Futur Oncol 7: 753-765.
- Ravikumar G, Ananthamurthy A (2014) Cyclin D1 expression in ductal carcinoma of the breast and its correlation with other prognostic parameters. J Cancer Res Ther 10: 671-675.
- McDonald ER, El-Deiry W (2009) Checkpoint genes in cancer. Annals of Medicine 33: 2001.
- Mukherjee S, Conrad SE (2005) c-Myc suppresses p21WAF1/CIP1 expression during estrogen signalling and antiestrogen resistance in human breast cancer cells. J Biol Chem 280: 17617-17625.
- Mohammadizadeh F, Hani M, Ranaee M, Bagheri M (2013) Role of cyclin D1 in breast carcinoma. J Res Med Sci 18: 1021-1025.
- Lebwohl ED, Muise-Helmericks R, Sepp-Lorenzino L, Serve S, Timaul M, et al. (1994) A truncated cyclin D1 gene encodes a stable mRNA in a human breast cancer cell line. Oncogene 9: 1925-1929.
- Khan N, Syed DN, Ahmad N, Mukhtar H (2013) Fisetin: A dietary antioxidant for health promotion. Antioxid Redox Signal 19: 151-162.
- Kenny FS, Hui R, Musgrove EA, Gee JM, Blamey RW, et al. (1999) Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in estrogen receptor-positive breast cancer. Clin Cancer Res 5: 2069-2076.
- Alle KM, Henshall SM, Field AS, Sutherland RL (1998) Cyclin D1 protein is overexpressed in hyperplasia and intraductal carcinoma of the breast. Clin Cancer Res 4: 847-854.
- Oyama T, Kashiwabara K, Yoshimoto K, Arnold A, Koerner F (1998) Frequent overexpression of the cyclin D1 oncogene in invasive lobular carcinoma of the breast. Cancer Res 58: 2876-2880.
- Baldini E, Camerini A, Sgambato A, Prochilo T, Capodanno A, et al. (2007) Cyclin A and E2F1 overexpression correlate with reduced disease-free survival in node-negative breast cancer patients. Anticancer Res 26: 4415-4421.
- Michalides R, Van Tinteren H, Balkenende A, Vermorken JB, Benraadt J, et al. (2002) Cyclin A is a prognostic indicator in early stage breast cancer with and without tamoxifen treatment. Br J Cancer 86: 402-408.
- Poikonen P, Sjöström J, Amini RM, Villman K, Ahlgren J (2005) Cyclin A as a marker for prognosis and chemotherapy response in advanced breast cancer. Br J Cancer 93: 515-519.
- Husdal A, Bukholm G, Bukholm IRK (2006) The prognostic value and overexpression of cyclin a is correlated with gene amplification of both cyclin A and cyclin E in breast cancer patient. Cell Oncol 28: 107-116.
- Donnellan R, Kleinschmidt I, Chetty R (2001) Cyclin E immunoexpression in breast ductal carcinoma: Pathologic correlations and prognostic implications. Hum Pathol 32: 89-94.
- Potemski P, Kusinska R, Watala C, Pluciennik E (2006) Cyclin E expression in breast cancer correlates with negative steroid receptor status, HER2 expression, tumor grade and proliferation. J Exp Clin Cancer Res 25: 59-64.
- Aaltonen K, Amini R-M, Heikkilä P, Aittomäki K, Tamminen A (2009) High cyclin B1 expression is associated with poor survival in breast cancer. Br J Cancer 100: 1055-1060.
- Bae SY, Lee JH, Bae JW, Jung SP (2020) Differences in prognosis by p53 expression after neoadjuvant chemotherapy in triple negative breast cancer. Ann Surg Treat Res 98: 291-298.
- Geradts J, Wilson P (1996) High frequency of aberrant p16(INK4A) expression in human breast cancer. Am J Pathol 149: 15-20.
- Arima Y, Hayashi N, Hayashi H (2012) Loss of p16 expression is associated with the stem cell characteristics of surface markers and therapeutic resistance in estrogen receptor-negative breast cancer. Intl J Cancer 130: 2568-2579.
- Göhring UJ, Schöndorf T, Kiecker VR, Becker M, Kurbacher C (1999) Immunohistochemical detection of H-ras protooncoprotein p21 indicates favorable prognosis in node-negative breast cancer patients. Tumor Biol 20: 173-183.
- Fayed YM (2012) Immunohistochemical expression of p27 in ductal carcinoma of breast and its correlation with HER2/neu expression and hormonal status. Egypt J Pathol 32: 33-41.
- Wang L, Wang R, Ye Z, Wang Y, Li X (2018) PVT1 affects EMT and cell proliferation and migration via regulating p21 in triple-negative breast cancer cells cultured with mature adipogenic medium. Acta Biochim Biophys Sin 50: 1211-1218.
- Pérez-Tenorio G, Berglund F, Merca AE, Nordenskjöld B, Rutqvist LE (2006) Cytoplasmic p21WAF1/CIP1 correlates with Akt activation and poor response to tamoxifen in breast cancer. Intl J Oncol 28: 1031-1042.
- Alkarain A, Jordan R, Slingerland J (2004) p27 Deregulation in breast cancer: Prognostic significance and implications for therapy. J Mammary Gland Biol Neoplasia 9: 67-80.
- Tsuchiya A, Zhang GJ, Kanno M (1999) Prognostic impact of cyclin-dependent kinase inhibitor p27kip1 in node-positive breast cancer. J Surg Oncol 70: 230-234.
- Maria K, Vassilis GG, George ZR, Petros L, Christos M (2002) High expression levels of p27 correlate with lymph node status in a subset of advanced invasive breast carcinomas: Relation to E-cadherin alterations, proliferative activity, and ploidy of the tumors. Cancer 94: 2454-2465.
- Newman L, Xia W, Yang HY, Sahin A, Bondy M (2001) Correlation of p27 protein expression with HER-2/neu expression in breast cancer. Mol Carcinog 30: 169-175.
- Deepu M, Daniel FR, Giorgio I, Anne ZJ, Joan C (2011) Loss of p27KIP1 expression in fully-staged node-negative breast cancer: Association with lack of hormone receptors in T1a/b, but not T1c infiltrative ductal carcinoma. Anticancer Res 12: 4401-4405.
- Leivonen M, Nordling S, Lundin J, Von Boguslawski K, Haglund C (2001) p27 expression correlates with short-term, but not with long-term prognosis in breast cancer. Breast Cancer Res Treat 67: 15-22.
- Abubakar M, Guo C, Koka H, Sung H, Shao N (2019) Clinicopathological and epidemiological significance of breast cancer subtype reclassification based on p53 immunohistochemical expression. NPJ Breast Cancer 5.
- Muhammad EMS, Ahmad AN, Guirguis MN, Ali A-E M (2012) Immunohistochemical p53 expression in breast carcinoma with correlation to clinico-pathological parameters. Med J Cairo Univ 80: 179-189.
- Nema S, Mehta P, Narang S (2019) Role of p53 and Ki-67 in prognostication of carcinoma breast. Indian J Pathol Oncol 6: 261-265.
- Lara-Gonzalez P, Westhorpe FG, Taylor SS (2012) The spindle assembly checkpoint. Current Biol 22: R966-R980.
- Marques S, Fonseca J, Silva PMM, Bousbaa H (2015) Targeting the spindle assembly checkpoint for breast cancer treatment. Current Cancer Drug Targets 15: 4.
- Wang Y, Jin F, Higgins R, McKnight K (2014) The current view for the silencing of the spindle assembly checkpoint. Cell Cycle 13: 1694-1701.
- Silva P, Barbosa J, Nascimento AV, Faria J, Reis R (2011) Monitoring the fidelity of mitotic chromosome segregation by the spindle assembly checkpoint. Cell Prolif 44: 391-400.
- Murray AW, Solomon MJ, Kirschner MW (1989) The role of cyclin synthesis and degradation in the control of maturation promoting factor activity. Nature 339: 280-286.
- Huang JN, Park I, Ellingson E, Littlepage LE, Pellman D (2001) Activity of the APC(Cdh1) form of the anaphase-promoting complex persists until S phase and prevents the premature expression of Cdc20p. J Cell Biol 154: 85-94.
- Zeng X, Xu WK, Lok TM, Ma HT, Randy YC (2019) Poon Imbalance of the spindle-assembly checkpoint promotes spindle poison mediated cytotoxicity with distinct kinetics. Cell Death and Disease 10: 314.
- Schuyler SC, Wu YF, Kuan VJ (2012) The Mad1-Mad2 balancing act--a damaged spindle checkpoint in chromosome instability and cancer. J Cell Sci 125: 4197-4206.
- Weaver BA, Cleveland DW (2006) Does aneuploidy cause cancer? Curr Opin Cell Biol 18: 658-667.
- Cohen-Sharir Y, McFarland JM, Abdusamad M, Marquis C, Bernhard SV (2021) Aneuploidy renders cancer cells vulnerable to mitotic checkpoint inhibition. Nature 590: 486-491.
- Marston AL, Wassmann K (2017) Multiple duties for spindle assembly checkpoint kinases in meiosis. Front Cell Dev Biol 5: 109.
- Maia ARR, de Man J (2015) Inhibition of the spindle assembly checkpoint kinase TTK enhances the efficacy of docetaxel in a triple-negative breast cancer model. Ann Oncol 26: 2180-92.
- Understanding targeted therapy. https://www.cancer.net/navigating-cancer-care/how-cancer-treated/personalized-and-targeted-therapies/understanding-targeted-therapy/
- Targeted cancer therapies, National Cancer Institute. https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies/targeted-therapies-fact-sheet/
- Targeted therapies, Memorial Sloan Kettering Cancer Center. https://www.mskcc.org/cancer-care/types/breast/treatment/systemic-therapy/targeted-therapies
- Targeted drug therapy for breast cancer. American Cancer Society. https://www.cancer.org/cancer/breast-cancer/treatment/targeted-therapy-for-breast-cancer.html/
- What is chemotherapy. https://www.healthline.com/health/breast-cancer/chemotherapy-for-her2-positive-breast-cancer#chemotherapy/
- Targeted therapy, National Breast Cancer Foundation, Inc. https://www.nationalbreastcancer.org/breast-cancer-targeted-therapy/
- Targeted cancer therapies, National Cancer Institute. https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies/targeted-therapies-fact-sheet/
- Targeted therapy to treat cancer, National Cancer Institute. https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies/
- Breast Cancer-Metastatic: Types of Treatment. Cancer.Net. https://www.cancer.net/cancer-types/breast-cancer-metastatic/types-treatment
- Yadav SS, Bhattacharya S (2021) Breast Cancer Scenario: A Review. Cancer Pages 2: 15.
- Visconti R, Monica RD, Grieco D (2016) Cell cycle checkpoint in cancer: a therapeutically targetable double-edged sword. J Exptl Clin Cancer Res 35: 153.
- Sun Y, Liu Y, Ma X, Hu H (2021) The influence of cell cycle regulation on chemotherapy. Intl J Mol Sci 22: 6923.
- Bhattacharya S (2021) Cytoskeleton and Epithelial Mesenchymal Transition. Cancer Pages 2: 16.
- Kotiyal S, Bhattacharya S (2014) Breast cancer stem cells, EMT and therapeutic targets. Biochem Biophys Res Comm 453: 112-116.
- Kotiyal S, Bhattacharya S (2015) Epithelial mesenchymal transition and vascular mimicry in breast cancer stem cells. Critical Reviews in Eukaryotic Gene Expression 25: 269-280.
- Kotiyal S, Bhattacharya S (2015) Lung cancer stem cells and their therapeutic targeting. Arch Stem Cell Res 2: 1009.
- Kotiyal S, Bhattacharya S (2016) Events of molecular changes in epithelial-mesenchymal transition. Critical Reviews in Eukaryotic Gene Expression. 26: 163-171.
- Collins K, Jacks T, Pavletich NP (1997) The cell cycle and cancer. Proc Natl Acad Sci USA 94: 2776-2778.
- Craig C, Wersto R, Kim M, Ohri E (1997) A recombinant adenovirus expressing p27 kip1 induces cell cycle arrest of loss of cyclin -CDK activity in human breast cancer cells. Oncogene 14: 2283-2289.
- Craig C, Kim M, Ohri E, Wersto R, Katayose D, et al. (1998) Effects of adenovirus-mediated p16INK4A expression on cell cycle arrest are determined by endogenous p16 and Rb status in human cancer cells. Oncogene 16: 265-272.
- Chen J, Willingham T, Shuford M, Bruce D, Rushing E, Smith Y, Nisen PD (1996) Effects of ectopic overexpression of p21(WAF1/CIP1) on aneuploidy and the malignant phenotype of human brain tumor cells. Oncogene 13: 1395-1403.
- Jin X, Nguyen D, Zhang WW, Kyritsis AP, Roth JA (1995) Cell cycle arrest and inhibition of tumor cell proliferation by the p16INK4 gene mediated by an adenovirus vector. Cancer Res 55: 3250-3253.
- Carnero A (2002) Targeting the cell cycle for cancer therapy. British J Cancer 87: 129-133.
- Schrump DS, Chen A, Consoli U (1996) Inhibition of lung cancer proliferation by antisense cyclin D. Cancer Gene Ther 3: 131-135.
- Kornmann M, Arber N, Korc M (1998) Inhibition of basal and mitogen stimulated pancreatic cancer cell growth by cyclin D1 antisense is associated with loss of tumorigenicity and potentiation of cytotoxicity to cisplatinum. J Clin Invest 101: 344-352.
- Galaktionov K, Beach D (1991) Specific activation of cdc25 tyrosine phosphatises by B-type cyclins: evidence for multiple roles of mitotic cyclins. Cell 67: 1181-1194.
- Garner-Hamrick PA, Fisher C (1998) Antisense phosphorothioate oligonucleotides specifically down-regulate cdc25B causing S-phase delay and persistent antiproliferative effects. Int J Cancer 76: 720-728.
- Bhattacharya S (2021) miRNA and cancer. In: G. Murdaca (Ed.), New Frontiers In Medicine And Medical Research, Book Publisher International, 15: 173-184.
- Otto T, Sicincki P (2017) Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer 17: 93-115.
- Sherr CJ, Bartek J (2017) Cell cycle-targeted cancer therapies. Annu Rev Cancer Biol 1: 41-57.
- Silva PM, Reis RM, Bolanos-Garcia VM, Florindo C, Tavares AA, Bousbaa H (2014) Dynein-dependent transport of spindle assembly checkpoint proteins off kinetochores toward spindle poles. FEBS Lett 588: 3265-3273.
- Targeted Therapies, Breastcancer.org https://www.breastcancer.org/treatment/targeted_therapies/
- Mathur P, Sathishkumar K, Chaturvedi M, Das P (2020) Cancer Statistics, 2020: Report From National Cancer Registry Programme, India. JCO Glob Oncol 6: 1063-1075.