High abundance proteins (HAP) were often removed from crude serum sample when finding biomarkers of diagnosing cancers, however, it will surely filtrate many potential biomarkers bound to HAP. In order to enable the detection of these potential biomarkers, saving the HAP will be of great significance for finding more biomarkers. Here, a serum proteomics technology was developed for finding biomarkers of liver-cancer from crude human serum without depletion of HAP. The crude human serum (CHS) was dispersed in a lysis buffer that has capacities for improving the protein resolution, and then separated with two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) for proteomic analysis. The differentially expressed proteins from Crude male serum (CMS) compared to crude serum of male patient with liver cancer (LCMPCS) were found and identified by a combined off-line approach of 2D-PAGE and matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF). Approximately 800 protein spots on a 2D-PAGE gel were detected by Melanie 4 software from samples both CMS and LCMPCS in the present of the lysis buffer. Significantly different expression of 24 proteins were detected in the LCMPCS compared to that in the CMS. Among these proteins, fifty were found up-regulated and eleven were found down-regulated. Most of these differently regulated proteins were identified by PMF and database search. These differential expression proteins were reported participating in some key pathway or biology process in cancer cells, indicating that they may present a biomarkers profile and may be helpful for liver-cancer diagnosis.
serum, biomarkers, liver cancer, proteomics, male
Although current largely reports confirmed that many significant biomarkers such as telomerase and ferritin from human serum have successfully been employed to diagnose various fatal diseases in clinic [1-3]. Most analytical approaches in the discovery and detection of these biomarkers showed a common disadvantage of the complicated experimental steps, the high fees, and the low reliability. Serum and plasma offer particularly promising resources for biomarkers discovery because collection of these samples in minimally invasive and the blood is thought to contain most of protein constitutes found in the body [4,5]. By comparing these differential proteins present in disease samples with those in control samples, it is possible to identify their expression change. These differences may be related to the response to organ toxicity or to risk factors preexisting before the acquisition of the disease [6]. There are recently been a tremendous in interest in applying mass spectrometry, 2D-PAGE, and HPLC on human plasma or serum for markers discovery and diseases diagnosis [1,7-9]. HAPs such as albumin, IgG, transferrin, antitrypsin make LAP difficult to be separated with 2D-PAGE approach from crude serum for proteomic analysis [10]. Several techniques such as action precipitation, gel filtration, ion-exchange chromatography, and reverse-phase chromatography have been applied to remove these HAPs, to enhance the detection sensitivity, and to increase efficiency of protein identification in plasma and serum analysis [1,8,11,12]. In addition, these techniques can only focus on enhancing sensitivity of LAP for MS analysis from human serum.
Recently a rapidly growing body of literature describes the use of proteome profiles generated by MALDI-TOF MS to classify serum samples based on disease states, particularly cancers [13,14]. Although 2D-PAGE is used routinely for proteome separation from human serum, but HAPs in the serum reduced the resolution of LAP in gel for protein identification strongly. To avoid many of the difficulties associated with gels, alternative gel-free separation is being developed which largely employs multi-dimensional HPLC [15], but this approach still shows much shortage such as the loss of significant proteins.
In this study, by using an off-line combination of both 2D-PAGE and MALDI-TOF-MS, we prepared a lysis buffer to improve the resolution of proteins separated with 2D-PAGE from CMS without depletion of HAP. We indicated that a 2D-PAGE approach describes hereto showed great advantage for finding clinic biomarkers from LCMPCS without HAP depletion, saving a lot of significant markers in a serum sample for MS identification.
Materials
In this study, blood samples of total 100 male volunteers were supplied by Zhongshan Hospital, Xiamen, China. The control blood samples of 50 males volunteers without obvious signs of disease, and another 50 LCMPCS samples diagnosed with various pathological pathways were mixed to eliminate differential proteins among individuals for proteome separation respectively before experiment. Each research had experiments repeated three times for making sure the results consistency under the same experimental conditions. Carrier ampholyte (Amersham Bioscience, USA), DTT, and CHAPS (Amresco, USA) were purchased. HCCA (INC, USA) and Trypsin (Promega, USA) was obtained. Other chemicals were purchased from Sigma- Aldrich (USA) unless otherwise specified.
Preparation of lysis buffer
The lysis buffer is composed of 7 M urea, 4% (w/v) CHAPS, 2 M thiourea, 60 mM DTT, 10 mM Tris, 1 mM EDTA, 0.2% (v/v) carrier ampholyte, 0.000 2% (w/v) bromophenol blue, which final total volume of 5 ml is strictly fixed before used.
Serum sample preparation
Total 100 blood samples both CMS and LCMPCS were employed to accomplish repeated research including data statistical analysis. Each experimental sample was allowed to coagulate for 30 min at 4℃ immediately and centrifuged for 10 min at 3,500 g in a swinging bucket centrifuge once the blood sample was collected from these volunteers. The supernatant fluid, serum, was a liquated and transferred into the new standard tube. The protocol ensures that the serum is collected and stored at room temperature for no more than 2 h before frozen at -70℃. The experimental samples that mixed 1 ml ice-cold serum samples with 9 ml lysis buffer, must be stored at -70℃ overnight, and then be thawed and centrifuged for 10 min at 6,000 g for removing thawless precipitate before used.
Approximately 5000±5 μg/ml serum proteins both control and experimental samples were adjusted by the lysis buffer for holding same protein concentration before separated by 2D-PAGE method.
2D-PAGE approach
A high-resolution 2D-PAGE method for separation of serum protein was carried out by an Investigator 2-D Gel System (Perkin-Elmer Co). The serum samples dissolved in the lysis buffer was loaded on the IEF strip with carrier ampholytes for first dimensional electrophoresis, which was performed at total 11,000 V·h in a stepwise fashion (0.25 h at 200 V, 0.5 h at 300 V, 1 h at 400 V, 16 h at 600 V). The prescription of the strip gel contains 0.55 g urea, 133 μl 30%T/3%C acrylamide, 200 μl 10% (v/v) NP-40, 200 μl DDW and 2.4% (v/v) carrier ampholytes. Degassed 20 mM NaOH served as catholyte, and 10 mM H3PO4 served as anolyte. SDS-PAGE for second dimensional electrophoresis was carried out, using an Investigator 2-D Gel System, at 15 mA per gel overnight at 10℃. Gels stained with sliver stained as currently described. Images from stained gels were digitalized at 300 dpi with a flatbed scanner (Perkin-Elmer Co.) and analyzed by using the Melanie 4 software for calculating protein spots on the SDS-PAGE gel. The protein digestion in gel was done with reference to the method as recently described by Qin, et al. [13]. The extracted peptides were dissolved in the 2.5 μl 0.5% (v/v) aqueous TFA for MS analysis.
Analysis of MALDI-TOF MS and PMF
Mass spectra of peptide mixtures were obtained by using a Reflex Ⅲ MALDI-TOF MS (Burker Daltonik, Bremen, Germany). α-Cyano-4-hydroxy cinnamic acid (HCCA) [50 mM HCCA in 4 vol. acetonitrile and 6 vol. 0.1% (v/v) TFA] served as matrix. The peptide spectra were recorded in the reflector mode and calibrated of acidic peptide [residues 26, M (H+) = 2961.5] and Angiotensin I (1296.5 Da) Accuracy of molecular ion mass was better than ±0.1 Da up to a mass of 6,000 Da [16,17]. PMF interrogations of appointed protein were performed by using MASCOT network. The maximum tolerance for masses was adjusted to be 40 ppm by an internal calibration that is autolysis products of trypsin where at most one missed cleavage for tryptic peptides was allowed. Mr and pI values of the analyzed spot were obtained from the 2D-PAGE gel. SWISS-PORT databases were used for significant proteins search and identification [10,13].
Direct separation from CMS
It was reported that the current methods can be effective in HAP remove, but many significant proteins bound to HAP might be lost from the crude serum once depletion of HAP [8,18].
Figure 1 shows protein separation in control CMS by 2D-PAGE with silver-stained in the broad pH range of 5.0-8.0 when applying 150±5 μg protein to a 2D-PAGE gel without depletion of HAP in advance. Figure 1 shows that many proteins of HAPs not only focused on the gel in the range of molecular weight around 68 kDa, but also covered with the spots of many LAPs. Image analysis with the Melanie 4 software indicated that approximately 120±10 protein spots were only detected from CMS samples (Figure 1). It is well seen that protein spots separated with 2D-PAGE approach directly was difficult to obtain high resolution of proteins from CMS sample without depletion of HAP by various tools of chromatography.
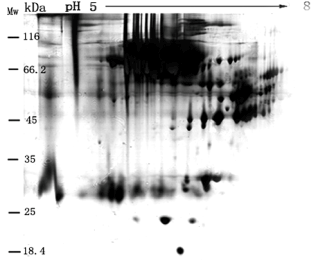
Figure 1. A 2D-PAGE image of proteome from CMS in the absence of lysis buffer. 150±5 μg protein content of CMS (1:9 ratio (v/v) of CMS to water) was directly loaded into a 2D-PAGE gel for protein separation without depletion of HAP. Image analysis with the Melanie 4 software indicated approximately 120 protein spots in the gel.
Role of a lysis buffer for protein separation
In order to enhance types of dissociative proteins, to reduce signal suppression effect of HAP, and to find more differential proteins, we prepared a lysis buffer to disperse polymeric proteins into single one from the CMS directly without depletion of HAP. A1:9 ratio (v/v) of CMS samples to the lysis buffer was mixed to separate protein by 2D-PAGE approach for MS analysis without depletion of HAP.
Figure 2 shows that approximately 800 protein spots were visualized by the Melanie 4 software from CMS sample in the present of lysis-buffer when applying 150±5 μg serum protein to a 2D-PAGE gel in the pH range 5.0-8.0. However, approximately 500 protein spots on the gel can be only identified by PMF and database search. In addition, 150±5 μg serum proteins for separation with 2D-PAGE approach is considered to be an optimized content of serum protein when applying protein contents of 100±5, 150±5 and 300±5 μg for comparison, respectively.
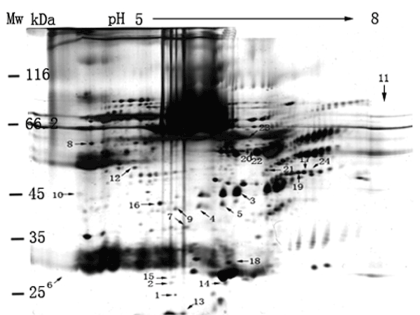
Figure 2. A 2D-PAGE image of proteome from CMS sample without depletion of HAP. 150±5 μg protein content of CMS (1:9 ratio (v/v) of CMS to the lysis buffer) was loaded into a 2D-PAGE gel for protein separation. Image analysis with the Melanie 4 software indicated approximately 800 protein spots in the gel.
The human plasma proteome is the largest representative of the human proteome present in any biological sample. HAPs such as human serum albumin (HAS) represented over 75% of all protein present in plasma and enhance signal suppression for MS analysis. The most common technique for removing these HAS from a sample need an essential tool of chromatography. Even so, many potential biomarkers bound to HAP might be filtrated by this chromatography such as ion-exchange and affinity approaches. Compared to Figure 1 results, Figure 2 results demonstrate that depletion of HAP from crude serum is not a necessary step for proteins separation by 2D-PAGE method although the several spots of HAP can still be observed in the gel. It indicates that the lysis buffer is able to not only disperse the interaction LAP and HAP for improving protein resolution in gel, but also save large numbers of LAP for finding its physiological function further. Compared to recently reports, we still found that the protein resolution including total spot numbers from human serum by 2D-PAGE approach describes here is better than those of other current methods with removal HAP. It is indicates that the lysis buffer plays an important role in driving to find more unknown significant proteins for disease diagnosing from CHM.
Marczynski reported that fragmentation changes of low molecular weight (LMW) –DNA can be found when blood cell (WBC) incubated with lysis buffer followed by constant-filed gel electrophoresis (CFGE) [18]. An improved lysis buffer for preparing bacterial whole-cell lysates for SDS-PAGE was reported [19]. These results indicated that the lysis buffer might play an important role in improving high resolution of proteins separated with 2D-PAGE from human serum. The results describe here indicate that the application of the lysis buffer has an advantage over the chromatography approach for proteome analysis from CHM, finding more novel significant biomarkers for disease clinic.
Differential proteins both CMS and LCMPCS
PMF can identify very small amount of protein in 2D-PAGE gels. The numbers of protein spots detected with sliver staining provided more than that with Commassie staining, but similar results with high quality PMF were still obtained when both analytical approaches were modified further.
Results in Figure 3 show the protein separation of LCMPCS sample dissolved with the lysis buffer by 2D-PAGE in a pH range from 5.0-8.0. The gel pattern (Figure 3) reveals approximately 800 protein spots when applying 150±5 μg serum protein to a 2D-PAGE gel. We can obtain the similar numbers of total protein spots both serum samples in 2D-PAGE gel. These results indicate that the application of lysis buffer in improving the protein resolution from the serum is successful.
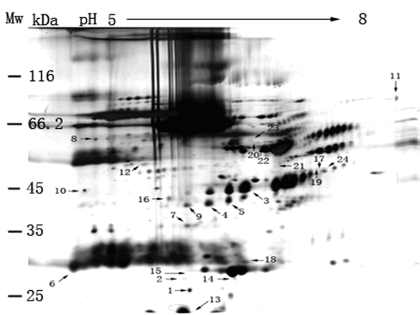
Figure 3. A 2D-PAGE image of proteome from LCMPCS sample without depletion of HAP. 150±5 μg protein content of LCMPCS (1:9 ratio (v/v) of LCMPCS to the lysis buffer) was loaded into a 2D-PAGE gel for protein separation. Image analysis with the Melanie 4 software indicated approximately 800 protein spots in the gel. Compared to the results in Figs.3 and 4, significant changes of 24 protein spots were observed.
Compared to a control sample (Figure 2), significant change of 24 protein spots were observed in the LCMPCS sample. These spots showing significant difference were further enlarged in order to analyze their biochemical parameters (Figure 4). Changes of spot numbers and intensity both control and experimental samples were quantified by high-resolution flatbed scanner and Melanie 4 software analysis (Materials and methods). Among these spots (Figure 4), seven (Figure 4a) were up–regulated, seven (Figure 4b) were down-regulated, eight (Figure 4c) showed low expression, and two (Figure 4d) showed high expression. All differential protein spots on the 2D-PAGE gel from both control and experimental samples were excised and digested with trypsinase.

Figure 4. Comparsion on enlarged spots of differential proteins from Figs 3 and 4 (A): Control protein spots from CMS; B: Experimental protein spots from LCMPCS. a: spots with up-regulated; b: spots with down-regulated; c: spots of low expression; d: spots of high experssion.
Differential protein identification
24 differential protein spots were subject to further identification in a combined off-line method of MALDI-TOF MS, PMF and database search, by an in gel trypsinase digestion approach. Among these 24 differential protein spots, seven have been primarily identified by PMF and database search. Of which certain biochemical parameters have been shown in the Figures 3 and 4 are summarized in Table 1 based on the results of database searches.
Table 1. Major differential expression proteins of LCPMS comparing with CMS
Spot |
Accession |
Score |
S.C.% |
Mr |
pI |
Descrition |
No.4 |
gi|37182794 |
73 |
14 |
45161 |
9.54 |
HS3ST3A1 |
No.7 |
gi|41202625 |
68 |
57 |
12210 |
8.66 |
tubulin beta D |
No.9 |
gi|62203472 |
62 |
28 |
24738 |
8.11 |
RAS oncogene family |
No.8 |
gi|563320 |
64 |
24 |
28141 |
5.39 |
apolipoprotein A-D |
No.12 |
gi|5815233 |
64 |
30 |
27602 |
7.68 |
60S acidic ribosomal protein PO |
No.15 |
gi|18027780 |
67 |
44 |
11783 |
9.28 |
unkown |
No.16 |
gi|18490206 |
75 |
20 |
38047 |
6.73 |
Cyclin H |
Based on a literature search, five differential proteins as biomarker for cancer clinic observed in Table 1 had been reported by previous scientists in details. These significant biomarkers are tubulin beta, RAS oncogene family, apolipoprotein A, acidic ribosomal protein, and Cyclin H [20-22]. For example, Hall, et al. reported that the presence of high levels of immunoreactive apoliporotein A-D (apo-D) in high grade prostatic intraepithelial neoplasia (HGPIN) and prostate cancer, but not in non- maligent epithelial cells. This result is consistent with HGPIN being an intermediate lesion in the transition to prostate cancer and suggests that cellular apoliporotein A-D expression is a marker of malignant transformation of the prostate [23]. Cdk-activating kinase (CAK) is a trimeric complex consisting of cdk7, cyclin H, and MAT1. Little is known about the regulation of the CAK complex though cyclin H [24,25]. Immunohistochemistry and real time quantitative polymerase chain reaction were used to determine cyclin expression and gene amplification in 219 tumors; the results indicated that cyclin H was overexpressed in all tumors [26-28]. These results mentioned above indicated that several biomarkers for cancer diagnosis can be found in a 2D-PAGE gel according to the approach describes hereto.
24 spots of significant differential protein were identified by proteomic techniques in this work. This series of experiments describes hereto demonstrates many advantages of direct proteomic analysis from CMS without depletion of HAP by using a combine off- line technology of 2D-PAGE and MALDI-TOF MS. These significant proteins in Table 1 were closely connected with the biomarkers for liver-cancer diagnosis. Although specific metabolic pathways for these proteins still cannot be elucidated fully, they are representative biomarkers produced in response to the disease of liver-cancers. Interestingly, these biomarkers (like fingerprints consisting of their molecular weights and IEF) can be located by their orientation on 2D-PAGE gels [13] and can be used to evaluate the level of the liver-cancer disease when applying a standard procedure for extracting and separating the proteins from the CMS and a scanner for reading the orientation of differential proteins in the gel.
2021 Copyright OAT. All rights reserv
This work was funded by the Science and Technology Project of Xiamen (No. 3502Z20143027).
- Ren Y, He QY, Fan J, Jones B, Zhou Y, et al. (2004) The use of proteomics in the discovery of serum biomarkers from patients with severe acute respiratory syndrome. Proteomics 4: 3477-3484. [Crossref]
- Yan L, Huang HQ, Jin HW, Wang QL (2004) Kinetics Study of Iron Release of Human Serum Ferritin with MALDI-TOF Mass Spectrometry and Electronic Spectrum Technology. Chem J Chinese Univ 25: 1889-1892.
- Schnabel CL, Steinig P, Schuberth HJ, Koy M, Wagner B, et al. (2015) Influences of age and sex on leukocytes of healthy horses and their ex vivo cytokine release. Vet Immunol Immunopathol 165: 64-74. [Crossref]
- Gadducci A, Cosio S, Carpi A, Nicolini A, Genazzani AR (2004) Serum tumor markers in the management of ovarian, endometrial and cervical cancer. Biomed Pharmacother 58: 24-38.
- Harper RG, Workman SR, Schuetzner S, Timperman AT, Sutton JN (2004) Low-molecular-weight human serum proteome using ultrafiltration, isoelectric focusing, and mass spectrometry. Electrophoresis 25: 1299-1306.
- Roy SM, Anderle M, Lin H, Becker CH (2004) Differential expression profiling of serum proteins and metabolites for biomarker discovery. Int J Mass Spectro 238: 163-171.
- Drukier AK, Ossetrova N, Schors E, Brown LR, Tomaszewski J, et al. (2005) Ultra-sensitive immunoassays using multi-photon-detection in diagnostic proteomics of blood. J Proteome Res 4: 2375-2378. [Crossref]
- Morris DL Jr, Sutton JN, Harper RG, Timperman AT (2004) Reversed-phase HPLC separation of human serum employing a novel saw-tooth gradient: toward multidimensional proteome analysis. J Proteome Res 3: 1149-1154. [Crossref]
- Anand S, Young S, Esplin MS, Peaden B, Tolley HD, et al. (2016) Detection and confirmation of serum lipid biomarkers for preeclampsia using direct infusion mass spectrometry. J Letipid Res 57: 687-696. [Crossref]
- Chromy BA, Gonzales AD, Perkins J, Choi MW, Corzett MH, et al. (2004) Proteomic analysis of human serum by two-dimensional differential gel electrophoresis after depletion of high-abundant proteins. J Proteome Res 3: 1120-1127. [Crossref]
- Wijte D, de Jong AL, Mol MA, van Baar BL, Heck AJ (2006) ProteomIQ blue, a potent post-stain for the visualization and subsequent mass spectrometry-based identification of fluorescent stained proteins on 2D-gels. J Proteome Res 5: 2033-2038. [Crossref]
- Petricoin EF, Liotta LA (2004) SELDI-TOF-based serum proteomic pattern diagnostics for early detection of cancer. Curr Opin Biotechnol 15: 24-30. [Crossref]
- Qin S, Ferdinand AS, Richie JP, O'Leary MP, Mok SC, et al. (2005) Chromatofocusing fractionation and two-dimensional difference gel electrophoresis for low abundance serum proteins. Proteomics 5: 3183-3192. [Crossref]
- Villanueva J, Shaffer DR, Philip J, Chaparro CA, Erdjument-Bromage H, et al. (2006) Differential exoprotease activities confer tumor-specific serum peptidome patterns. J Clin Invest 116: 271-284. [Crossref]
- Wang MZ, Howard B, Campa MJ, Patz EF Jr, Fitzgerald MC (2003) Analysis of human serum proteins by liquid phase isoelectric focusing and matrix-assisted laser desorption/ionization-mass spectrometry. Proteomics 3: 1661-1666. [Crossref]
- Zhu JY, Huang HQ, Bao XD, Lin QM, Cai Z (2006) Acute toxicity profile of cadmium revealed by proteomics in brain tissue of Paralichthys olivaceus: potential role of transferrin in cadmium toxicity. Aquat Toxicol 78: 127-135. [Crossref]
- Chen DS, Huang HQ, Wu HZ, Cai ZW (2006) Optimized Separation and Identification of Proteome from the Baccal Ganalion of Aplysia (Notarcus leachii cirrosus Stimpson). Chem J Chinese Univ 27: 1257-1261.
- Marczynski B, Kraus T, Rozynek P, Schlösser S, Raithel HJ, et al. (2001) Changes in low molecular weight DNA fragmentation in white blood cells of workers highly exposed to asbestos. Int Arch Occup Environ Health 74: 315-324. [Crossref]
- Chen M, Christen P (1997) Removal of chromosomal DNA by Mg2+ in the lysis buffer: an improved lysis protocol for preparing Escherichia coli whole-cell lysates for sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Anal Biochem 246: 263-264.
- Chen YY, Lin SY, Yeh YY, Hsiao HH, Wu CY, et al. (2005) A modified protein precipitation procedure for efficient removal of albumin from serum. Electrophoresis 26: 2117-2127. [Crossref]
- Hasegawa S, Miyoshi Y, Egawa C, Ishitobi M, Tamaki Y, et al. (2002) Mutational analysis of the class I beta-tubulin gene in human breast cancer. Int J Cancer 101: 46-51. [Crossref]
- Kavallaris M, Kuo DY, Burkhart CA, Regl DL, Norris MD, et al. (1997) Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J Clin Invest 100: 1282-1293. [Crossref]
- Prasannan L, Misek DE, Hinderer R, Michon J, Geiger JD, et al. (2000) Identification of beta-tubulin isoforms as tumor antigens in neuroblastoma. Clin Cancer Res 6: 3949-3956. [Crossref]
- Hall RE, Horsfall DJ, Stahl J, Vivekanandan S, Ricciardelli C, et al. (2004) Apolipoprotein-D: a novel cellular marker for HGPIN and prostate cancer. Prostate 58: 103-108. [Crossref]
- Krempler A, Kartarius S, Günther J, Montenarh M (2005) Cyclin H is targeted to the nucleus by C-terminal nuclear localization sequences. Cell Mol Life Sci 62: 1379-1387. [Crossref]
- Waters NC, Woodard CL, Prigge ST (2000) Cyclin H activation and drug susceptibility of the Pfmrk cyclin dependent protein kinase from Plasmodium falciparum. Mol Biochem Parasitol 107: 45-55.
- Bondi J, Husdal A, Bukholm G, Nesland JM, Bakka A, et al. (2005) Expression and gene amplification of primary (A, B1, D1, D3, and E) and secondary (C and H) cyclins in colon adenocarcinomas and correlation with patient outcome. J Clin Pathol 58: 509-514. [Crossref]
- Romagnoli S, Fasoli E, Vaira V, Falleni M, Pellegrini C, et al. (2009) Identification of potential therapeutic targets in malignant mesothelioma using cell-cycle gene expression analysis. Am J Pathol 174: 762-770. [Crossref]