Acute kidney injury (AKI) is a global public health concern, impacting nearly 13.3 million patients and resulting in three million deaths per year. Chronic kidney disease (CKD) has increased by 135% since 1990, representing the pathology with the fastest growth rate worldwide. The annual costs of dialysis and kidney transplants range between US$35,000 and US$100,000 per patient. Despite its great impact, kidney disease has remained mostly asymptomatic for many years. AKI continues to be a major, unmet medical condition for which there are no pharmacological treatments available, while animal models are limited to provide direction for therapeutic translation into humans. Currently, serum creatinine is the standard biomarker to identify nephrotoxicity; however, it is a late stage biomarker. Hence, there is a pressing need to study in vitro biomarkers for the assessment of nephrotoxicity in order to develop new and safer drugs. Understanding of the mechanisms by which molecules produce nephrotoxicity is vital in order to both prevent adversity and treat kidney injury. In this review, we address new technologies and models that may be used to identify earlier biomarkers and pathways involved in nephrotoxicity, such as cell culture, omic approaches, bioinformatics platform, CRISPR/Cas9 genome-editing, in silico, organoids and 3D bioprinting, considering adverse outcome pathways (AOP).
acute kidney injury (AKI), adverse outcome pathways (AOP), chronic kidney disease (CKD), early diagnosis, nephrotoxicity models, new technologies
Kidney disease is a relatively silent disease and, in most cases, only becomes apparent to those suffering from it in the later stages of the illness. This can be partially attributed to the fact that most people are not aware of the early signs associated with kidney disease, coupled with the fact that these signs are not always recognized by healthcare providers [1]. Nonetheless, it is estimated that, annually, around three million deaths worldwide are caused by acute kidney injury (AKI). However, these numbers may be underestimated, given that half of the patients admitted to intensive care units develop AKI during hospitalization, and 27% of these die before being released from the hospital. It is also well-known that these figures constitute only a small proportion of the total number of reported cases [2].
AKI is an expected event that occurs relatively quickly, such as a few hours or days, after organ failure. The full process is still not clear. AKI is characterized by a reduction in the glomerular filtration rate (GFR), which leads to an increase in serum creatinine (sCr) or blood urea nitrogen (BUN). However, measuring BUN levels is not a reliable factor to predict AKI [3].
The frequency of AKI has significantly increased in recent decades and is now recognized as a significant cause of Chronic Kidney Disease (CKD) and kidney failure in general [4]. AKI is characterized as kidney disease that persists up to 90 days; this disease is considered chronic CKD when it surpasses 90 days [2,5].
Between 19% and 26% of the cases of AKI among hospitalized patients are caused by medication-induced adverse effects that injure the kidney [6]. AKI is still a major, unmet medical problem for which no efficient pharmacological treatments exist [7]. Therefore, an understanding of the mechanisms by which molecules may cause harm (such as those leading to AKI) is vital in order to both prevent toxicity and treat kidney injury.
Acknowledging the need for action, an increasing number of global advocacy initiatives, such as World Kidney Day, the International Society of Nephrology 0by25, and the Lancet Kidney Campaign aim to raise public awareness regarding the consequences, costs, and impact of both CKD and AKI [2].
Acute kidney injury
AKI is a worldwide public health concern, impacting nearly 13.3 million patients per year [7]. AKI is associated with high morbidity and mortality - over 1.7 million deaths per year among adults and children worldwide [8]. AKI is disseminated throughout 72 countries and is higher in developing countries. The mortality rates of critically ill patients with AKI in hospitals ranges between 20% and 70% [9,10]. In the United States alone, half a million hospital admissions each year are complicated by AKI, resulting in total costs of approximately $10 billion [11]. Worldwide, only half of the individuals who need renal replacement therapy can be treated, due to difficulties related to access to care or lack of diagnosis. In fact, estimates suggest that 2.5 to 5 million patients are not properly treated [12]. In addition, the costs of dialysis and kidney transplants range between $35,000 and $100,000 per patient annually.
Prescription drugs and other traditional low-level medication is the major cause of AKI, resulting in nearly 25% of all patients who use at least one nephrotoxic drug. The other 75% of the cases stem from other etiologies. However, the main cause of mortality and morbidity by AKI can be explained by drug-induced nephrotoxicity in over 20% of hospital-acquired AKIs [13-16]. Despite this high rate, data show that only 7% of new drug candidates fail in preclinical trials because of nephrotoxicity [17], whereas the incidence of patients in intensive care units developing AKI is about 30-50% [18]. This discrepancy may help to explain the underestimation of nephrotoxicity in preclinical trials [19]. Therefore, AKI is still a major medical condition for which no pharmacological treatment has been developed [20-22].
The incidence of AKI has greatly increased over the past two decades, and it is now recognized as an important cause of CKD and kidney failure [4,23]. However, the causal pathway leading to CKD is complex and will require a broad multidisciplinary response [24]. Fortunately, the kidney carries an extremely regenerative potential and, in some cases, can be completely recovered ‘restitutio at integrum’ after an AKI. Nevertheless, there is strong evidence suggesting that repair can be incomplete, and an episode of AKI may lead to CKD. The appearance of tubular response after AKI will shed light on the connection between incomplete repair and future risk of CKD [3]. Thus, the ‘AKI to CKD transition’ might involve incomplete renal tubule repair after AKI, eventually triggering interstitial renal fibrosis, making it critical to investigate the cells responsible for normal repair. Hence, by developing new targeted therapies to reduce injury and promote healing in AKI, it will be possible to determine which cells should be targeted.
In clinical practice, the early detection of AKI has had no impact on clinical decisions. In addition, therapeutic possibilities for AKI are very limited, and it is unclear if there is any benefit in either early or late onset of renal replacement therapy. As a result, the early or late detection of AKI, in most cases, does not impact the clinical course of the patient. To some extent, this may explain why most biomarkers have not been included in the panel of classic laboratory parameters used in the clinical setting. Hence, advances are needed in this area in order to understand the gap related to renal disease.
Chronic kidney disease
According to the “Kidney Disease: Improving Global Outcomes (KDIGO)” guidelines, CKD is characterized by abnormalities of kidney structure or function that are present for more than 3 months [5].
There are few markers, such as neutrophil gelatinase-associated lipocalin (NGAL), kidney injury molecule-1 (KIM-1), and nephronectin (NPNT), as well as the validated markers of proteinuria and microalbuminuria, that can detect the progression of AKI into CKD, which makes it an important area for investigation [25,26]. Among those who survive the long-term outcomes of AKI, patients with CKD can progress into the end-stages of renal disease (ESRD) or into the aggravation of pre-existing CKD, which can quickly lead to ESRD [12,23,27].
Thus, AKI, which was previously thought to have no major consequences for recovered patients, may lead to poor life quality as well as long-term financial costs [28], which increases the need for public awareness regarding the early signs of the disease and investment in further research [26]. CKD represent an important risk factor for cardiovascular and cerebrovascular diseases and progress toward ESRD [29,30], which can only be treated by renal replacement therapies such as hemodialysis, peritoneal dialysis, or transplants [30,31]. Kidney failure is the most severe form of CKD and is fatal if not treated by dialysis or kidney transplant. Because of the uncertainty of what causes CKD, many patients do not receive the correct treatment [4,32].
The resources required to treat CKD-associated complications (including kidney failure) impose a huge burden in such countries as Malaysia, Tunisia, Turkey, Chile, Mexico, and Uruguay [33]. In fact, it has been shown that the burden of CKD is on the rise, as indicated by the increase in both deaths and ESRD [4,23,34-36].
Nevertheless, CKD is not on the priority list of non-communicable diseases, and only a few countries have explicit policies or public programs focused on preventing and controlling CKD. This unfortunate scenario is of concern because there is a gap between recent advances in treatment and the increase in mortality worldwide, thus creating barriers that limit the progress of basic research that would ultimately lead to new treatments (translational research type 1) [37].
CKD represents the pathology with the fastest growing incidence worldwide [38]. Its incidence has virtually duplicated between 1990 to 2010 [39]. The global prevalence of CKD is estimated at around 10% to 15%, with similar estimates in men and women, and is more frequent in high-income countries. The impaired renal function is caused by diabetes mellitus in 30-40% of the cases, hypertension in 20%, inflammation of the glomeruli (glomerulonephritis) in 13%, interstitial nephritis in 10%, and polycystic kidney disease in 6%. Regardless of the cause, the common final outcome of almost all progressive CKDs is renal fibrosis [40,41].
The actual incidence of renal disease seems to be underestimated because many deaths attributed to diabetes have actually been caused by renal disease [42]. Moreover, hypertension and obesity are among the growing non-communicable diseases and represent important risk factors for CKD [23]. Thus, identifying CKD has become imperative not only to prevent the progression of the disease, but also to reduce the risk of cardiovascular morbidity and mortality. Hence, new biomarkers are paramount for early identification of the disease.
Biomarkers for nephrotoxicity
Currently, AKI diagnosis is based on serum creatinine’s rise and/or fall in urine production. These are not renal-specific markers and have major limitations. Alternatively, there are two sets of biomarkers available for clinical use: NGAL and the combination of urinary insulin-like growth factor-binding protein 7 (IGFBP-7) and tissue inhibitor of metalloproteinases 2 (TIMP-2), two markers of cell-cycle arrest. These biomarkers seem to be specific and sensitive enough to be used together with additional markers for AKI to better stratify renal injury [43].
In 2008, the Food and Drug Administration (FDA) designated seven biomarkers of nephrotoxicity to test in animals and, on a case-by-case basis, to be used in humans. These included urinary KIM-1, β2-microglobulin, cystatin C, clusterin, trefoil factor 3, albumin, and total protein. These markers, among others (eg, urinary NGAL, urinary IL-18, and L-FABP - liver fatty acid binding protein), have been evaluated in many conditions but have rendered inconclusive results as a predictive model. In most cases, the data obtained with biomarkers tested in animals are compared to changes in sCr concentrations in humans, but results are questionable. In 2014, the NephroCheck test, which helps to determine if critically ill patients are at risk of developing moderate-to-severe AKI following 12-hour testing, was approved by the FDA. However, it is unknown at which time-point the measurement should be performed to ideally predict AKI [26,44,45]. Thus, there are few markers for progression of AKI to CKD. Moreover, this strategy of using biomarkers also serves to determine a specific point in the course of AKI when the predictive value of the biomarker should be tested [11].
AKI is an extremely heterogeneous and complex condition. Even though there is an effort to identify AKI biomarkers that could be used for risk assessment, diagnosis, severity, and/or outcome of AKI with high predictive power, an ideal, universal AKI marker is unlikely to be discovered. Thus, considering the heterogeneous nature of AKI, a complex multimodal approach, including a detailed risk assessment and the implementation of new biomarkers, is advisable to prevent and manage AKI [46].
For this reason, several biomarkers that comprise the different aspects of AKI are required. Sensitive detection of an acute reduction in kidney function and injury in various renal structures, as well as the evaluation of the degree of renal injury, are the types of information provided by marker panels. Ideally, these markers should also be mediators of other disease pathways in AKI. Even better, these markers are the key to understanding the pathological mechanisms, to indicating the etiology of AKI, to providing references for future therapies, as well as to following therapy progress. Early biomarkers to estimate renal tissue damage is an advance that replaces the old low-sensitive methods for diagnosis by using better AKI criteria [7,47].
AKI biomarkers may be used to reveal higher sCr levels after ischemia reperfusion injury (IRI) and can also be used an early indication of the progression from AKI to CKD. This may also indicate that the process used was not as effective as intended. For instance, fibrosis is still found in patients, despite the reacquisition of normal biochemical parameters, such as plasma creatinine removal; progression to CKD is still the most frequent result [48].
KIM-1 is a type I cell membrane glycoprotein with extracell immunoglobulin and mucin domains [49-51]. The mRNA and protein for KIM-1 are expressed at very low levels in the normal rodent kidney; however, expressions increase dramatically after an injury in the proximal tubule epithelial cells in postischemic rodent kidney and in humans during ischemic acute renal ups and downs of using KIM-1, and according to Medić, et al. [52], larger trials are still necessary before KIM-1 can be tested in broader clinical trials. Although KIM-1 is a sensitive and specific marker for kidney tubular injury, it is difficult to measure it in acute settings [53].
By contrast, Ichimura, et al. [54] demonstrated that human KIM-1 exhibits homology to HAVCR1, and studies using gentamicin [55,56], amphotericin B [57], and cisplatin (no publishing data) have shown that the gene HAVCR1 (KIM1) might be used as in vitro biomarkers of nephrotoxicity.
To find new drugs for AKI and prevent its transition into CKD, it is crucial to better understand how tubular injury and repair work. Once this knowledge is acquired, it would be possible to use translational biomarkers to assess drug targets and their pathways. Furthermore, the expected biological effect of the drug can be revealed by specific biomarkers, i.e., whether the drug has reached the pathophysiological pathway for which it was developed and what is the expected clinical outcome [3,47,48].
Animal model
Animal models have been widely used to understand the pathogenesis and underlying mechanisms of renal disease. As shown in the Figure. 1, mice and rats are the most commonly used to study nephrotoxicity and therapeutic targets, as well as to identify new biomarkers, mainly because these are easily bred and are relatively inexpensive to house and maintain [58]. Nonetheless, few advances have been achieved in understanding nephrotoxicity mechanisms or in identifying either a new biomarker or a panel of biomarkers. According to our search, monkeys are also used; however, regardless of the size and breed of the animals, results did not show a good predictability to identify adverse effect in humans.
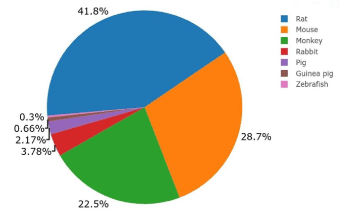
Figure 1. Proportion of animals used in in vivo studies in renal toxicity
In fact, the failure of existing animal models to accurately predict nephrotoxicity has been a major barrier to the development of safer drugs. Extensively studied for several decades, animal models of human AKI have, to date, yielded no specific therapy that benefits the human disease, either in preventing its occurrence, ameliorating its severity, hastening its recovery, or retarding the risk of ensuing CKD. Such inability to translate these models’ uncovered salutary strategies into therapies for human AKI has led to a questioning of the value of these models: specifically, their resemblance and fidelity to human AKI. Overlapping this kidney-centered discussion are broader debates in relevant biomedical communities, national as well as international, regarding a worrisome, relative lack of congruence of conclusions in preclinical studies and the slow pace of translating findings obtained in animal models into new therapies for human disease [59].
Ischemia–reperfusion is one of the main causes of AKI; by definition, human AKI implies decreased GFR. Hence, conventional AKI models are mainly the result of kidney ischemia–reperfusion, exposure to endogenous or exogenous toxins and urinary tract obstruction [60]. However, this may not be the case for animal models if only one kidney is injured. Hence, despite the abundant literature on the pathogenesis of ischemia–reperfusion, no novel therapy has been able to be applied in clinical scenarios [61].
Exogenous drugs or poisons, as well as endogenous toxins, are widely used to stimulate AKI for their side or poisoning effects. Among these models, 6-20 mg/kg cisplatin can result in acute tubular injury within 72 h, whereas the administration of 40-200 mg/kg gentamicin in rats for 4–10 days can induce acute renal failure [62].
Chemotherapy comprised three cycles of high dose (80-100 mg/m2) cisplatin administered on days 1, 22, and 43. Cisplatin is a drug widely used for chemotherapy (80-100 mg/m2 – IV administered on days 1, 22, and 43) to effectively treat various cancers and gentamicin is an aminoglycoside antibiotic (6-12 g/day, IV administered, for 2-6 weeks) commonly used to prevent gram-negative bacterial infection. However, both drugs have limitations in clinical practice due to nephrotoxicity. For many years, nephropathy caused by cisplatin and gentamicin have been widely studied, but it is still unclear, although different models using different animals have been used and many hypotheses have been suggested [63-66]. Nevertheless, these hypotheses do not fully reproduce human clinical diseases. Hence, we do believe new in vitro strategies are necessary to study the molecular mechanism involved in nephropathy so as to reach proper clinical diagnoses and implement effective therapeutic interventions.
Actually, some AKI models in mice have been used with both advantages and disadvantages (Table. 1). Even so, models tend to have a high clinical relevance, for example, classical models contain a high knowledge background and are technically simple and reproducible. The types of models are incomplete and many details, such as model techniques and modeling time, are not mentioned. In addition, the majority of these have a variable response between models, expected acute renal necrosis is not always achieved, AKI is not produced clinically and pathologically, surgery is required, reproducible outcome is dependent on accurate Ischemia/Reperfusion time, and the renal function can be compensated by a non-ligated kidney [62].
Table 1. Advantages and limitations of conventional AKI animals models
Model |
Renal target |
Design |
Advantages |
Limitations |
Isquemia-reperfusion |
Proximal tubule and endothelium |
Clamping time:
30-45 min
Reperfusion time:
24-48 hours |
Pathology similar to human disease |
Requires surgery |
Drug induced |
Cisplatin |
Proximal tubule |
Sinlge IP injection
(6-20 mg/kg)
72 hours |
Pathology, timing, and drug doses similar to human disease |
No clinical correlate |
Gentamicin |
Proximal tubule and glomerulus |
Serial IP injections
(40-200 mg/kg/day)
4-10 days |
Induces rapidly progressive CKD |
No fully reproduce human clinical diseases |
Aristolochic acid |
Proximal tubule |
Serial IP injections
(5 mg/kg/day)
5 days |
Induces rapidly progressive CKD |
No clinical correlate |
Folic acid |
Proximal tubule |
Single IP injection
(250 mg/kg)
24-48 hours |
Pathology findings similar to human AKI from other causes |
No clinical correlate |
Glycerol |
Proximal tubule |
Single IM injection
(8 mL/kg of glycerol 50%)
24-48 hours |
Clinically relevant, resembling human rhabdomyolysis |
Severe and focuses in the symptoms, not the mechanism |
Warfarin |
Proximal tubule and glomerulus |
5/6 nephrectomy for 3 weeks and 8 days on warfarin |
Useful to study AKI caused by anticoagulants |
Not guarantee the same mechanism |
Abbreviations: IP: Intraperitoneal; IM: Intramuscular.
Aristolochic acid (AA) and high dose folic acid (FA) are also frequently used to study AKI-CKD transition, with AKI models developed by warfarin and glycerol [62]. Nephrotoxicity caused by AA was first reported in Belgium in a group of patients who ingested weight loss pills containing extracts of Chinese herbal powdered roots [67]. The findings in humans are supported by laboratory animal studies showing that oral exposure to AA caused tumors in the urinary tracts of various animal species. Although this model showed good results it did not work for other drugs. In 2001, the FDA advised consumers to stop using any products that may contain AA, after seeing an increase in nephropathy, or kidney disease, among users [68].
Fink, et al. [69] demonstrated that single intravenous doses of FA in rats causes direct nephrotoxic effects; hence, it has been used to study AKI-CKD transition. Some studies have used intraperitoneal injection [60,70,71]. However, FA is the synthesized form of folate present in fortified foods and supplements, such as grains, and has a higher bioavailability than naturally occurring folate [72].
Thus, AA and FA are used at low doses for oral administrations in human and at high dosages and intravenous and peritoneal doses when studying nephrotoxicity in animals. Hence, AA and FA do not represent good models, since the profiles for causing nephrotoxicity are completely different.
Warfarin (WRN) therapy can result in AKI by causing glomerular hemorrhage and renal tubular obstruction by red blood cell (RBC) casts [73]. Analysis of more than 15,000 warfarin-treated patients showed that WRN affects approximately 33% of CKD patients and 16% of non-CKD patients who experienced an international normalized ratio >3.0. This study also identified that mortality rates in patients with WRN was significantly higher than in patients without WRN [74]. However, excessive anticoagulation in control animals was not associated with changes in creatinine levels, and renal morphology was normal. Therefore, Ware, et al. [75] developed a new method to study nephrotoxicity using an excessive amount of warfarin combined with a 5/6-nephrectomy model. Although this method could cause functional and morphological damage of the remnant kidney, it does serve to demonstrate the effect. Nevertheless, it does not guarantee the same molecular mechanism responsible for nephrotoxicity, which is the main element to be discovered.
AKI is also a common complication of rhabdomyolysis and accounts for the high mortality [62,76,77]. To reproduce the typical symptoms observed in humans, rats or mice are deprived of water for 24 h, after which a 8-10 mL/kg dose of 50% glycerol is administrated in the hindlimb muscle [78,79]. We do not believe glycerol could be a good model, given that the model is focused on the symptoms, which is not the main point of our study. In addition, glycerol is used in human food at low doses. Thus, it does not represent a good model to study nephrotoxicity.
Mechanisms of disease generation and progression in AKI and CKD have yet to be fully understood [80,81]. Although several clinical studies have investigated early stage predictive biomarkers of kidney disease, few have been applied in clinical practice [82]. CKD models can include mainly renal mass reduction, diabetic nephropathy, hypertension glomerular injury, IgA nephropathy, polycystic kidney disease, and chronic tubulointerstitial nephritis (Table. 2).
Table 2. Advantages and limitations of experimental CKD animal models
Pathology |
Model |
Transition AKI-CKD |
Advantages |
Limitations |
Comments |
Renal mass reduction |
5/6 nephrectomy (rats) |
Depends on the mice strains |
Equivalent to humans |
Two surgery requires. The second one having a high mortality rate, especially in mice, and requiring technical expertise; mice strains are differentially responsive to renal mass reduction with respect to CKD progression; it is irreversible and the amount of kidney left to study is small and may be distorted by surgical procedure |
No clinical translation of successful |
Diabetic Nephropathy |
Streptozotocin mice/rats; NOD mice BB-DP rat; ob/ob mice db/db mice; STZ-eNOS-/-; db/db-eNOS/mice |
No |
Gene modified; commercially available; available on multiple strains; useful to study mechanisms of diabetic nephropathy. Reproduce human physiopathology |
None of the many animal models perfectly replicate the human disease; expensive; some strains are infertile; cause modest albuminuria without decreased GFR |
No clinical translation of successful |
Hypertensive Nephropathy |
SHR rats+UNX; angiotensin II infusion models |
Depends on the dose/time |
Highly relevant to hypertension nephropathy; useful to study AngII effect over kidney |
SHR rats are more resistant to streptozotocin-induced diabetes; uninephrectomy is required to promote significant kidney injury; no progressive GFR loss; higher dose or long exposure time periods increase serum creatinine levels; high cost; slow progression |
No clinical translation of successful; long exposure time periods stress animals |
Acquired Glomerular Injury |
Focal segmental Glomerulosclerosis: adriamycin (rat, mice) or puromycin (rat) |
Depends on the strains |
Mimic acute glocmerular injury |
Usually no adequate reproduction of slowly progressive human disease |
No clinical translation of successful NOD |
IgA Nephropathy |
ddY mouse, HIGA mice; Uteroblobin-deficient mice |
|
Reproduces human pathology |
Mild disease development usually without progression towards end-stage renal disease; no progressive GFR decrease |
No clinical translation of successful |
Polycistic Disease |
Genetically engineered mouse model |
Yes, but no specific to renal disease |
Useful to study PKD mechanisms and to find therapeutic targets; new insights into the molecular mechanisms of cystogenesis and associated progression of CKD; resulted in regulatory approval for Tolvaptan in Japan for human ADPKD |
Mouse models usually lack the variety of human phenotypes |
ARPKD mouse models usually lack the variety of human phenotypes |
Chronic Tubulointersticial Nephritis |
Simple, dose-dependent decreased GFR; Possibly reversible (rats) |
Model no specific for human |
Simple, dose-dependent decreased GFR; possibly reversible |
Adenine-ged male rats had a more severe decline in kidney; models CKD extrarenal complications; function and molecular changes |
Adenine intoxication is not an issue in human |
Hereditary Glomerular Injury |
Pkd1 or Pkd2 gene engineered; Col4a43 gene knockout mouse; Alport syndrome |
Depends on the strains |
Reproduce features of human disease, including progressive GFR loss; develop proteinuria and renal failure |
Difficulty to translation of genetic findings into improved patient care; high costs |
Resulted in clinical recommendations |
Stone Disease |
Natural development (dogs and cats) |
No information |
Without external factors to cause disease |
Few studies |
Long-term |
Abbreviations: GFR: glomerular filtration rate; SHR: Spontaneously hypertensive rat; BB-DP: BioBreeding-diabetes prone; NOD: nonobese diabetic; ob/ob: obese; db/db mouse: type 2 diabetes; DBA/2J: model for congenital experimental glaucoma; STZ: streptozotocin;eNOS: Endothelial nitric oxide synthase; UNX: Uninephrectomy; ddY mouse: outbread; HIGA: Nephropathy with Hyperserum IgA; PKD: polycystic kidney disease; COL: collagen genes
Renal mass reduction (remnant kidney model) is one of the most widely used models to study CKD. Subtotal (5/6) nephrectomy has been a mainstay of studies of progressive CKD. The right kidney is removed and the upper and lower poles (2/3 of the left kidney) are resected after ligation of the left renal artery. After surgery, activation of the renin-angiotensin system (RAS) can cause glomerular hypertension/hyperfiltration [61,62,83,84].
Glomerular injury may be acquired or hereditary. Acquired diabetic nephropathy (DN) is the leading cause of ESRD. There are many rodent models for diabetic nephropathy; however, none of them perfectly mimics the human disease [85]. The Animal Models of Diabetic Complications Consortium (AMDCC) defines the ideal rodent model of human DN and complications [86,87]. Several models of gene targeting have reproduced mutations to generate murine models of hereditary podocyte-based nephropathies, including focal segmental glomerulosclerosis and Alport syndrome [61]. Hypertension-induced renal injury is usually investigated by using spontaneous hypertension. Additionally, uninephrectomy is required to promote significant kidney injury in the form of albuminuria without reduced GFR [88]. Chronic exposure to pressure-increasing doses of angiotensin II in rats and mice caused renal dysfunction [89].
Focal segmental glomerulosclerosis (FSGS) is a common primary glomerular injury, characterized mainly by podocyte loss, glomerular fibrosis, and marked proteinuria [90]. Although there is currently no primary FSGS model available, several secondary FSGS models have been established. Non-genetic models mimic acute glomerular injury but not the spontaneous slowly progressive FSGS observed in the clinic. Experimental podocyte depletion, using adriamycin or puromycin aminonucleoside, in turn leading to CKD, emphasizes the key role of podocytes in the preservation of the renal function [61,91].
IgA nephropathy (IgAN) is the most common human glomerulonephritis, but no specific therapy has been discovered. However, none of these models enabled the identification of the factors that control the transition between disease onset and progression to ESRD. Thus, the underlying mechanism of IgAN is still not fully understood [61,62].
Polycystic kidney disease (PKD) includes a group of human monogenic disorders inherited in an autosomal dominant (ADPKD) or recessive (ARPKD) fashion. Genetically engineered mouse models, in which disease-causing genes were targeted, now mimic human ADPKD, ARPKD, and atypical PKD. Those models have already provided new insights into the molecular mechanisms of cystogenesis and the associated progression of CKD. However, ARPKD mouse models usually lack the variety of human phenotypes [61].
Primary chronic tubulointerstitial nephritis is associated with an immune-mediated infiltration of the kidney interstitium by inflammatory cells, which may progress to fibrosis. This is characterized by tubular atrophy, interstitial fibrosis, and interstitial inflammation in the absence of significant glomerular disease. Most animal models of chronic tubulointerstitial nephritis are secondary to ischemia, proteinuria, AKI, or other primary kidney diseases [61,92]. Thus, the processes involved are different and do not demonstrate similar mechanisms; hence, it does not represent a good model.
In addition to the animals already mentioned dogs and cats also have been suggested to study the calcium oxalate stone formation, considering that a naturally occurring animal model could be more useful. This strategy could represent a good model; however, the pathologic and etiologic mechanism surrounding calcium oxalate nephrolithiasis is incomplete [93].
Thus, despite the valuable new insights into kidney disease gained from existing models, many do not fully reproduce human clinical diseases. In the AKI, models are not stable and reproducible, while in the CKD, the disease is quite complex [62]. However, in addition to each model's specific limitations, all animal-model research into human diseases is ultimately restricted by the need to translate findings across species. This calls for the wider use of human-based models to complement and reduce the use of experimental in vivo research [94].
In this scenario, we do believe that new strategies and models without animals are required to better mimic processes involved in AKI and CKD. These models must mainly address molecular mechanisms, since the mere understanding of them may help to develop therapeutic interventions and clinical diagnoses, as well as to identify new biomarkers for nephrotoxicity. Hence, we also believe that new strategies are paramount for advances in this area. Moreover, alternative methods, such as those addressed in this review, represent important tools that can shift the paradigm of nephropathy and the way new, more effective, and safer molecules can be developed.
In vitro model
In vitro toxicity studies, whether cell or tissue, have been used to study the mechanisms of action and toxic effects of drugs in order to assess efficacy and safety in humans regarding exposure to xenobiotics, and may also help to classify them according to their toxic capacity. The knowledge of the mechanism of toxic action involving a xenobiotic is of paramount importance, since it provides significant aid in the evaluation of predictive values, in addition to improving the selection of new drugs to treat specific pathologies [95-97].
The number of in vivo studies has grown proportionally to in vitro studies (Figure. 2) according to our search. Despite this, such models have demonstrated limitations in generating knowledge of nephrotoxic processes in humans. By contrast, there is an increasing demand for new methods that refine, reduce, and replace animal use. Cell culture techniques are extremely relevant tools for in vitro studies of nephrotoxicity. The number of studies with animal models and in vitro renal toxicity has grown in the last 20 years. Despite this increase of both strategies, no reported results have provided a better understanding of nephrotoxicity mechanisms.
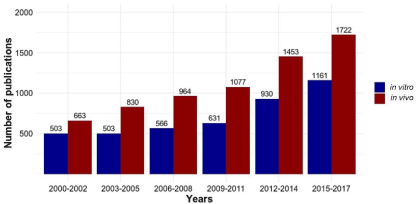
Figure 2. Number of in vivo and in vitro studies in renal toxicity
It is difficult to study specific epithelial cell types because the renal cortex carries a heterogeneous population of renal tubule segments. Fortunately, improvements in the methods used to grow homogeneous cultures of kidney cells allow one to access proper cells over different portions of the nephron. Other possibilities are the 3D printing of scaffolds to test the efficacy of new drugs, which will be different in scale and throughput from the assays used in the initial toxicological screening of chemicals [98].
Regardless of these advances, the kidneys produce very complex structures with blood filtration and urine re-absorption units needed for their function and homeostasis. It will also be highly important to consider the quality control of kidney organoids for disease modeling, added to the potential sources for developing kidney regenerative therapies. Conversely, there is currently no consensus regarding the methods that should be used for the evaluation of the quality of kidney organoids, mainly in terms of their functional characterization [99].
Recently, the mouse ENCODE Consortium reported that, while there are many similarities between human and mouse genomes, there are important differences as well. Consequently, animals are not good models for nephrotoxic effects in humans. Many discrepancies in DNA and differences in gene expression patterns have been found, potentially restricting the usefulness of some disease models based on mice [100]. More than 160 inherited genetic kidney diseases are now known.
Alternatively, by establishing human induced pluripotent stem cells (hiPSCs) from patients with a given genetic disease, such as degenerative disorders and cancer, it might be possible to study customized disease mechanisms and to perform drug screening in vitro instead of using animal models. Most importantly, updated advances in genome editing have provided new approaches to modeling genetic kidney diseases by using human pluripotent stem cells (hPSCs) in vitro [101–104].
In addition, the market potential for NATs (Non-Animal Tests) is enormous. Approximately $100 billion was spent on R&D in 2014 by the top 25 pharmaceutical companies (based on recorded global sales for 2014) and a recent survey of the members of Pharmaceutical Research and Manufacturers of America estimated that more than 20% of total R&D expenditure was on preclinical research [98,105].
In the long term, there is the potential to use NAMs (Non-Animal Models) to support the development of personalized medicines through the use of human-based approaches that will enable the identification of the possibility for side effects caused by drugs, or variations of efficacy in the population. The selection of patients on the basis of their predicted response allows a drug to continue through its development into clinical use in circumstances where its progress would otherwise have been suspended. This could bring enormous benefits to both patients and companies [98].
In addition, there is an increasing demand for new methods which refine, reduce, and replace animal use. Cell culture techniques are extremely relevant tools for in vitro studies of nephrotoxicity. The number of studies with animal models and in vitro renal toxicity has grown in the last 20 years. And despite this increase in both strategies, it has not resulted in a better understanding of nephrotoxicity mechanisms (Figure. 2).
While in vitro models have helped us to understand nephrotoxicity mechanisms, it is paramount to use new technologies, such as omic approaches (transcriptomics, proteomic, and metabolomics), bioinformatics platforms, CRISPR/Cas9 genome-editing and in silico modeling to advance this process. As shown in Figure. 3, these new technologies have been increasingly applied to study renal toxicity.
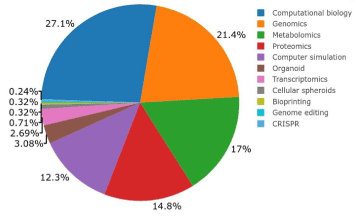
Figure 3. New technologies used to study renal toxicity
Omics technologies
Omics technologies include transcriptomics, proteomics, and metabolomics and are methodologies applied for biomarker identification useful in understanding the mechanisms of nephrotoxicity. Such technologies offer unbiased approaches to identify new biomarkers of AKI. Circulating or urinary microRNAs are being evaluated, and kidney-specific DNA methylation patterns are also being analyzed. Researchers have also been paying attention to urinary extra cell vesicles, which contain mRNA, microRNA, and proteins that mimic the cell physiology and pathophysiology along the nephron. The components of urinary extracellular vesicles are a source of information about disease pathogenesis, and they may serve as diagnostic and prognostic biomarkers, since they may enable target identification for drug discovery or provide evidence of biological activity in response to therapeutic agents. Among other objectives, omics studies seek to identify those that are mainly descriptive in nature, through the integration of experimental approaches and new computational modeling, such as in systems genetics, which will be generated in order to help identify marker sets to guide diagnoses, monitor disease progression, and identify new therapeutic targets [8,20,106].
The European Consortium for High Throughput Research in Rare Kidney Diseases (EURenOmics) has been working on a cohesive bioinformatics platform to study rare nephropathies. Additionally, a renal phenomenon database is being created, using the Human Phenotype Ontology website. The phenotype information will be linked to genomic, transcriptomic, proteomic, and metabolomics studies, omics datasets, and the public domain knowledge-base in a systems biology approach to identify molecular pathways associated with phenotypic features [107].
This situation thus becomes a great opportunity to advance in vitro studies for renal disease by analyzing gene expression. A bioinformatics platform will be an important tool to identify biomarkers that could be paramount in developing new medicines as well as in identifying nephrotoxicity and stopping the processes of AKI and CKD. Bioinformatics platforms are able to respond to such challenges as renal disease.
Transcriptomics: Studies on transcriptional profiling have revealed molecular markers and potential regulatory pathways of renal repair. A few key markers become active on the developmental pathways that have been reported during repair. By using high-resolution technologies, such as RNA sequencing and translational profiling specific to cell compartments within the kidney, further insights into injury and repair processes will be gained through the study of repairing transcriptome and cell-specific translatome. An enhanced understanding holds promise for both the identification of modern therapeutic targets and biomarker-based evaluation of the damage-repair process [20].
Both proximal and distal tubular epithelial cells mount an acute transcriptional response to IRI. The earliest genes to be induced after in vivo injury (within 4 h after injury) include Fos, Jun, and Egr1 [108]. Fos is induced predominantly in the thick ascending limb (TAL) [109]. The latter observation suggests that the distal tubule, in addition to the proximal tubule, also senses the acute insertion. Subsequent microarray-based gene expression profiling studies encountered a similar immediate-early response, including Fos and Egr1, after IRI [110,111]. It is possible to identify toxicity on proximal and distal tubules, which will create an enormous advantage. Consequently, gene expression studies using tubular and renal cells may help to identify earlier nephrotoxicity [20].
Adler, et al. [112] showed that primary human proximal tubular epithelial cells (HPTEC) are a good in vitro model for screening for renal toxicity because they have differentiated epithelial cell characteristics. In addition, HO-1 expression proved to be a better option than currently used to predict toxicity in HPTEC, especially when combined with the total number of cells in the same assay. In addition, the approach used by the authors can be used to screen a large number of compounds besides can be used in combination with existing cytotoxicity assays.
The understanding of how the several cell types in the kidney communicate, in order to regulate the intrinsic repair mechanisms and their contribution to post injury fibrosis, remains a substantial challenge. Nonetheless, a continued focus is still placed on cell-type–specific signatures and a broadening of analysis beyond early injury, which in turn makes it feasible to provide important new insights. Clearly, the major clinical objective is to develop new analytical tools to diagnose both short- and long-term outcomes, as well as to develop new therapeutic strategies to improve existing repair processes and reduce the long-term risk of CKD after AKI [103]. In sum, all of these strategies could help to develop new agents to treat renal disease.
Proteomics: Proteomics is considered one of the most promising ‘omics techniques for biomarker discovery in AKI, as it may identify specific molecular targets for early injury in different renal compartments. Almost all human diseases are characterized by a complex panorama at the molecular level, and it is imperative to acquire a global proteome picture to depict pathways and proteins with pivotal roles in pathogenesis. Proteomics studies have indicated that a single biomarker cannot fully account for the complexity of human diseases, and thus it is preferential to use biomarker panels.
Hence, it is thought that proteomics is a good tool for discovering and identifying new premature biomarkers. The main target of proteomics is to characterize the information flow within the cell and the organism, through protein pathways, interactions and networks, thereby allowing a hypothesis-free identification of disease biomarkers [113]. The search for biomarkers using urinary proteomics approaches has proven to be effective in CKD, diabetic nephropathy, AKI, ureteropelvic junction obstruction, vesicoureteral reflux, renal Fanconi syndrome, acute renal allograft rejection, cancer, DN, chronic rhinosinusitis, atherosclerosis and calcific aortic valve disease, and posterior urethral valves [106,114-119].
Metabolomics: Metabolomics is defined as the analysis of molecules smaller than 1,000 Da, which are transformed as a result of, and in support of, an organism’s metabolism. The metabolome accounts for a complete set of metabolites that can be produced and consumed by organisms [117,120]. The results of metabolomics experiments constitute big data and require sophisticated data handling strategies and advanced statistical tools. In addition, before any statistical analysis, data preprocessing must take place to ensure the best possible results from the applied statistics [106]. Spectral alignment, normalization, transformation, and scaling are indispensable stages in metabolomic data preprocessing. Data normalization is required for accurate metabolite quantification. To normalize data, a common method is based on endogenous metabolites, such as urinary creatinine. However, creatinine itself may be somewhat variable, and subject to variation owing to diseases such as AKI [106,120,121].
The potential to identify drug targets from metabolomics will be amplified by the coordination of efforts to analyze samples of human kidney tissue and other biomaterial, such as urine and blood, by using state-of-the-art genomic, proteomic, and metabolomic approaches, together with detailed patient phenotyping and the use of existing biomarkers to discover and qualify new therapeutic purposes. Genetic data should be linked to existing phenotypic information or generated from customized human tissue models, by using induced pluripotent stem cells and targeted mutation, followed by differentiation to human kidney tissue. To give support to these efforts, enhanced disease prototypes are needed to reflect the complexity of CKD [117,122].
Equally challenging is the multifactorial origin of AKI and the fact that the changes of molecular expression induced by AKI are difficult to distinguish from those of the diseases associated with or causing AKI, such as shock or sepsis [106]. In the past, these experiments were focused on a single metabolite that was attributed to a specific disease or enzymatic reaction [123]. Nowadays, such technologies as GC/MS (gas chromatography-mass spectrometry) and computational tools allow for a more extensive and wide-ranging investigation of many metabolites within a single measurement, providing a broader insight into mechanisms of diseases [106]. For this reason, it is very important to employ a panel of biomarkers.
New and cutting-edge techniques have been, and continue to be, developed; the challenge with the present status of metabolomics is its transition to clinical use. Integrating metabolomic data with other omics data for the purpose of drug discovery, and development is one direction in which this field may proceed [124,125].
The low sensitivity of current clinical markers (sCr and BUN) in early stages of the development of acute kidney injury limits their utility. Rapid LC/MS (liquid chromatography-mass spectrometry)-based metabolic profiling of serum was demonstrated in a key study showing that metabolomics could provide new indicators of AKI. Currently, omics technologies provide a great opportunity for research in rare renal diseases, as it is possible to probe the diseased organ directly [126]. Hence, we believe it is important to compare results obtained from patients and to evaluate them using cells, mainly because there are several different types of renal disease. This would thus be a safer strategy for using clinical results as a starting point.
CRISPR/Cas9 genome-editing
The fast advancement of genome-editing technologies has provided new pathways in the study of the genetic basis of human diseases and in the development of targeted therapeutic strategies that would not have been possible with traditional pharmacological drugs. Chemical toxicants act by many different mechanisms, however, the genes involved in adverse outcome pathways (AOPs) and AOP networks are not yet characterized. Functional genomic approaches can reveal both toxicity pathways and susceptibility genes, through knockdown or knockout of all non-essential genes in a cell of interest, and the identification of genes associated with a toxicity phenotype following toxicant exposure [127-129].
In this environment, new strategies could improve the engineering of genome-editing tools, and appropriate regulatory practices, together with genome editing, will accelerate discoveries in basic science and clinical translation, which will in turn aid in the process of identifying new biomarkers to become more robust. Hence, advances in genome editing with CRISPR/Cas9 will promote the use of kidney organoids to study inherited genetic kidney diseases [127].
The potential use of kidney organoids derived from hPSC combined with genome engineering technologies, particularly the CRISPR/Cas9, is a novel approach for the targeted modification of the renal epigenome to study renal organ dysfunction, such as CKD [130]. In addition, this technology could be further used to produce kidney phenotypes.
In silico
In silico, or computational, approaches to predicting and identifying compounds associated with kidney disease are based on the use of existing data and the ability to draw chemistry-based inferences from those data [131]. The concepts are relatively trivial in that, should data of acceptable quality be available for a compound, then there is no need for further testing. If no suitable data are available for a compound, then inferences can be made from it, eg, making an analogy with similar compounds or identifying chemical/molecular features that may be indicative of the ability to cause disease. All of these resources together form a battery of computational approaches, or even of a compound, that can shed light on the disease-causing potential [132]. The general availability of data related to the ability of a compound to cause damage to the kidney has been reviewed by Pletz, et al. [133]. The resources can be classified according to the type of data they represent. The most relevant for human risk assessment are those for the effects of compounds in humans; these are available as part of clinical trial data or from reports of adverse drug reactions. Websites, such as www.clinicaltrials.gov, are useful to retrieve such data. While potentially relevant, they can only be used for the drugs to which they relate and lack consistency between clinical trials. To develop and evaluate in silico models, the results of animal tests done in the past are now useful, particularly low-dose chronic toxicity testing that show the superior predicting power of in silico models. Several resources have been compiled of repeated dose toxicity values and are freely available. These become useful in understanding and evaluating effects to the kidney when they record organ-level effects. The COSMOS database was developed specifically to achieve this and can be searched for organ-level toxicity, including effects to the kidney and bladder [134]. Other similar databases of repeated-dose toxicity data include REPDOSE [135] and HESS [136]. Other significant data resources of repeated-dose toxicity values that may require licensing and/or payment include Chemtunes (MN-AM, https://www.mn-am.com/products/chemtunes), Vitic Nexus (Lhasa Ltd, https://www.lhasalimited.org/products/vitic-nexus.htm), and LeadScope Toxicity Database (Leadscope Inc, http://www.leadscope.com/product_info.php?products_id=78). It is important to note that while these data resources may shed light on which compounds may promote disease, or damage, to the kidney, they were not developed specifically for this purpose.
There are fewer databases for in vitro data. However, one important resource that will aid in understanding, from the mechanistic point of view, is the compilation of the United States Tox21 initiative. Tox21 set about to screen large numbers of compounds (currently over 10,000) with high content assays, some of which are relevant to the effects and mechanisms of kidney disease and damage [137]. This immense and freely accessible resource is available through the US EPA’s Chemistry Dashboard [138]. The Chemistry Dashboard is a cutting-edge and rapidly evolving resource for in silico assessment. It enables a user to input a chemical structure, retrieve existing (in vivo and Tox21) data, make predictions, and build knowledge about a substance. It is currently focused on a number of environmental and human health effects and could be applied to provide information on kidney disease if used appropriately, i e, the data can assist in the identification and rationalization of chemicals associated with disease. Another important in silico tool that brings together data through the development of chemistry-based groups is the OECD (Organization for Economic Co-operation and Development) QSAR (Quantitative Structure-Activity Relationship) Toolbox (https://www.qsartoolbox.org/). Designed for grouping and read-across to allow for data gap filling, the Toolbox includes access to mechanistically based profilers to find similar molecules.
One of the most powerful methods to use existing data to make predictions for compounds with no data (the aforementioned process of data gap filling) is to form groups of similar compounds and “read-across” data from one molecule to another [139]. The use of the read-across paradigm has seen growth in recent years, most notably due to its application in the European Union’s Registration, Evaluation, Authorization and restriction of Chemicals (REACH) legislation [140]. The identification of similar compounds is a broadly applied concept and requires a certain basis. One approach has been to use markers of organ-level toxicity to define similarity [141]. What seems particularly relevant here are the chemistry-based structural alerts or biomarkers for kidney disease. Thus, should compounds be associated with the same chemistry alerts or biomarkers, then similarity in activity may be assumed, ie, a prediction of harm can be made by analogy to the similar substance. While conceptually simple, there are numerous caveats to the approach and care must be taken [142]. One means of providing relevance from the in silico side is to ensure that the alerts are anchored on a mechanistic basis and specifically to relevant AOPs (see section below).
The logical identification of structure-activity relationships related to kidney disease, ie, the sub-molecular features intimately associated with damage, has enabled the concepts of structural alerts to be defined. Such alerts are easily coded into software. Some structural alerts for kidney damage are available in the literature; for example, Myshkin, et al. [143] published alerts for a number of effects. Efforts are currently underway to rationalize the small number of publicly available structural alerts for harm caused to the kidney and to add new alerts where they are missing. This work is being supported by a compilation of over 200 compounds known to cause harm to the kidney [133], which have assisted in the evaluation of existing alerts and will enable efficient screening of new molecules for these effects.
Historically there have been many efforts to form quantitative associations between chemical structure and biological activity – the development of the so-called QSARs [131]. It is fundamental that QSARs require suitable data for development and, ideally, should be mechanistically based. As a result, particularly of the need for robust data sets, there have been relatively few QSAR models developed for nephrotoxicity; available models have been reviewed by Pletz, et al. [133]. For instance, Lei, et al. [144] used machine learning approaches to develop models for damage to the urinary tract. While some progress has been made, the development of QSARs for the full suite of harmful effects to the kidney is limited at the current time, but it can be expected to improve due to the better access to reliable data for modeling.
Using HPTEC and a set of 44 compounds, subdivided into HPTEC-toxic, non-HPTEC-toxic and non-nephrotoxic compounds, Su, et al. [145] described an approach based on high-throughput imaging, phenotypic profiling and machine learning methods to predict human nephrotoxicity. This approach does not require characterization of injury mechanisms and is capable of identifying structurally distinct xenobiotic-induced nephrotoxicity that lead similar phenotypic endpoints as from six sets of nuclear and actin cytoeskeletal features with high efficiency and accuracy.
In addition to using information from chemistry for modeling, according to Soo, et al. [146], integration of information obtained from in vitro models into computational algorithms that incorporate patient-specific physiological parameters will not only reduce late drug loss from the development pipeline, but also facilitate the development of safer drugs and improve compound management of clinically important adverse nephrotoxic effects.
Stem cells
Stem cell therapy is an innovative approach to ameliorate IRI due to its antioxidative, immunomodulatory, and anti-apoptotic properties. However, the majority of the studies are confined to experimental animal models, although, several studies have investigated mesenchymal stem cells-based therapies for both acute and chronic kidney disease in clinical trials. Hence, it is paramount to understand the biological effects and mechanisms of action of stem cell therapy to improve its therapeutic benefits. In addition, more translational studies are needed to provide a more comprehensive understanding of stem cell-based therapies and to ensure their safety for future clinical applications [147,148].
New models such as organoids and 3D bioprinting also are important tools which may help us to understand nephrotoxicity mechanisms.
Organoids
Nephrons are complex architectural 3D structures. Hence, to recapitulate these structures ex vivo, the development of 3D culture systems is required. Organoids are 3D organ-like tissues that mimic in vivo organs structurally and functionally in culture plates. Kidney organoids derived from hPSCs are an attractive approach for studying mechanisms of human inherited kidney diseases, which might be applied to more common diseases as well as to the development of new drugs using human tissues, which may facilitate the translation of the findings obtained with this methodology to humans. However, to optimize this approach, differentiation protocols, genetic background, and epigenetic variation need to be considered in depth when disease phenotypes are analyzed in kidney organoids [99].
One goal of hPSC studies is to regenerate the kidney function. Kidneys form complex structures with blood filtration, and the urine re-absorption unit is paramount for their functioning and homeostasis. There are many challenges in the use of organoids to generate functional bioengineered kidney tissues. Vascularization of kidney organoids needs to be induced in an organized way to direct blood flow from arteries in order to drain into venous structures. This represents one of the great challenges related to vascularization [99].
3D bioprintin
In addition, the use of 3D bioprinting has proven to be an option to study nephrotoxicity, namely a functional bioengineered kidney with vasculature, multiple cell types, which include proximal tubular cells, and an extracell matrix. A new system is being used to generate heterogeneous structures in 3D that enable the perfusion of liquid into lumens lined with vascular or tubular cells. It may be employed to mimic blood flow and intratubular flow in vascular and tubular channels [99,149,150].
Adverse outcome pathways
The AOP framework is a systematic, transparent approach used to organize existing toxicological knowledge and translate mechanistic information to adverse effects at higher levels of organization based on causal relationships between endpoints (Figure. 4). As such, the AOP framework has been proposed as a means to relate alternative types of data (in silico, in vitro and in vivo, biomarker-type data) to endpoints of concern to chemical risk assessors (eg, survival, growth, and reproduction). This application in the regulatory field is timely given the current momentum to move toxicology from broadly empirical in vivo assessments of unique chemical effects to predictive approaches that make use of newer tools and alternative data [151-153].
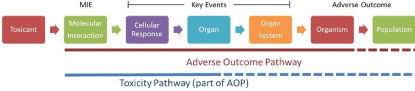
Figure 4. Adverse outcome pathways Adverse outcome pathways. Adapted from Browne P, Noyes PD, Casey WM, Dix DJ (2017) Application of Adverse Outcome Pathways to U.S. EPA’s Endocrine Disruptor Screening Program. Environ Health Perspect 125: 096001
With reference to AOPs for renal disease, a small number of studies have been performed using molecular initiating events (MIEs) to identify the following AO: adenomas and carcinomas, and renal failure and mortality, respectively. Most authors do not inform about the progress of the studies (https://aopwiki.org/aops).
Hence, it is paramount to use new tools in order to develop renal AOPs. Some studies have been performed utilizing biochemistry [154–156] and genomics [55-57]. However, it is important to make these data public through database, like the Comparative Toxicogenomics Database (CTD), in an attempt to achieve faster and more reliable results.
Thus, in terms of using the biomarker data, the AOP paradigm is a useful starting point. There are many advantages to the use of AOPs, including the rationalization of response and the ability to form the basis of quantitative AOPs, dissemination through the AOP-wiki, as well as the elucidation of the MIE [141]. In addition, the use of the MIE of an AOP is a robust means of developing structural alerts and has been applied for other organ-level toxicities [141,157]. At the time of writing this chapter, structural alerts for nephrotoxicity that allow for grouping and read-across are still under development [133].
It is clear that AOP will greatly help reduce animals in research involving nephrotoxicity, but it also uncovers the molecular mechanisms that may speed up the discovery of biomarkers for disease staging and therapy individualization, as well as the design and testing of novel therapeutic strategies that are safer to be developed, together with diagnoses and more accurate treatments.
The failure of existing animal models to accurately predict nephrotoxicity has been a major barrier to the development of safer drugs. The failure has mainly been due to the complexity of the physiology of the kidney, coupled with the variety and mechanisms of potential adverse effects, which animals are not able to identify. The application of advances in the understanding of molecular signaling and function would be a great benefit to find a means through which to prevent toxicity and/or treat kidney injury. Thus, new tools have been implemented, using alternative methods in order to elucidate mechanisms of nephrotoxicity. The use of in vitro models is at the forefront of the understanding of functional maturity and injury responses caused by drugs. However, it is essential to integrate existing in vitro methods with new technologies. For instance, organoids, 3D bioprinting, omics (transcriptomics, proteomics, and metabolomics), bioinformatics platforms, CRISPR/Cas9 genome-editing, and in silico modeling have been proposed as potential new tools to understand and identify nephrotoxicants. The main goal of the use of new and existing technologies will be to generate an integrated model in which new biomarker(s) and/or pathway(s) involved in nephrotoxicity can be established as early as possible. The use of AOPs is helping to organize the information from the new technologies and the Molecular Initiating Event is expected to represent a robust means to detect toxicity. Therefore, combining a panel of biomarkers may improve the performance of the interpretation and predictability of nephrotoxicity. Gene expression integrated with computational analysis is also a key area that will facilitate the understanding of nephrotoxicity mechanisms and will consequently aid in the development of safer new drugs and be useful in the detection and monitoring of adverse effects from nephrotoxicants.
We thank the institutions that promote Brazilian research National Council for Scientific and Technological Development (CNPq), Coordination for the Improvement of Higher Education Personnel (CAPES), Research Support Foundation of Minas Gerais State (Fapemig) and Pró-Reitoria de Pesquisa da UFMG (PRPq/UFMG).
Humane Society International
The author reports no conflicts of interest in this work.
- MacLeod A (2009) NCEPOD report on acute kidney injury—must do better. Lancet 374: 1405-1406.
- Kellum JA, Ronco C, Bellomo R (2016) Acute kidney disease and the community. Lancet 387: 1974-1976.
- Kramann R, Kusaba T, Humphreys BD (2015) Who regenerates the kidney tubule? Nephrol Dial Transplant 30: 903-910.
- Levin A, Tonelli M, Bonventre J, Coresh J, Donner JA, et al. (2017) Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet 390: 1888-1917.
- KDIGO CKD Work Group (2013) KDIGO 2012 Clinical practice guideline for evaluation & management of CKD. Kidney Int 3: 5-14.
- Murugan R, Karajala-Subramanyam V, Lee M, Yende S, Kong L, et al. (2010) Acute kidney injury in non-severe pneumonia is associated with an increased immune response and lower survival. Kidney Int 77: 527-535.
- Mehta RL, Cerdá J, Burdmann EA, Tonelli M, García-García G, et al. (2015). International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet 385: 2616-2643.
- Ho J, Dart A, Rigatto C (2014) Proteomics in acute kidney injury - Current status and future promise. Pediatr Nephrol 29:163-171.
- Fortrie G, De Geus HRH, Betjes MGH (2019) The aftermath of acute kidney injury: A narrative review of long-term mortality and renal function. Crit Care 23: 1-11.
- Schmitt R, Coca S, Kanbay M, Tinetti ME, Cantley LG, et al. (2008) Recovery of kidney function after acute kidney injury in the elderly: A systematic review and meta-analysis. Am J Kidney Dis 52: 262-271.
- Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW (2005) Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16: 3365-3370.
- Liyanage T, Ninomiya T, Jha V, Neal B, Patrice HM, et al. (2015) Worldwide access to treatment for end-stage kidney disease: A systematic review. Lancet 385: 1975-1982.
- Bellomo R (2006) The epidemiology of acute renal failure: 1975 Versus 2005. Curr Opin Crit Care 12: 557-560.
- Kaufman J, Dhakal M, Patel B, Hamburger R (1991). Community-acquired acute renal failure. Am J Kidney Dis 17: 191-198.
- Nash K, Hafeez A, Hou S (2002) Hospital-acquired renal insufficiency. Am J Kidney Dis 39: 930-936.
- Wonnacott A, Meran S, Amphlett B, Talabani B, Phillips A (2014) Epidemiology and outcomes in community-acquired versus hospital-acquired aki. Clin J Am Soc Nephrol 9:1007-1014.
- Frost, Sullivan Rang HP (2007) Drug discovery and development. Churchill Livingstone: Elsevier.
- Devarajan P (2011) Biomarkers for the early detection of acute kidney injury. Curr Opin Pediatr 23: 194-200.
- Fuchs TC, Hewitt P (2011) Biomarkers for drug-induced renal damage and nephrotoxicity—An overview for applied toxicology. AAPS J 13: 615-631.
- Kumar S, Liu J, McMahon AP (2014) Defining the renal repair transcriptome after acute kidney injury. Semin Nephrol 34: 404-417.
- Lameire NH, Bagga A, Cruz D, De Maeseneer J, Endre Z, et al. (2013) Acute kidney injury: an increasing global concern. Lancet 382: 170-179.
- Sales GTM, Foresto RD (2020) Drug-induced nephrotoxicity. Rev Assoc Med Bras 66: s82-s90.
- Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, et al. (2013) Chronic kidney disease: global dimension and perspectives. Lancet 382: 260-272.
- Horton R, Berman P (2015) Eliminating acute kidney injury by 2025: an achievable goal. Lancet 385: 2551-2552.
- Kashani K, Kellum JA (2015) Novel biomarkers indicating repair or progression after acute kidney injury. Curr Opin Nephrol Hypertens 24: 21-27.
- Zuk A, Bonventre JV (2016) Acute kidney injury. Annu Rev Med 67: 293-307.
- Waheed S, Matsushita K, Astor BC, Hoogeveen RC, Ballantyne C, et al. (2013) Combined association of creatinine, albuminuria, and cystatin C with all-cause mortality and cardiovascular and kidney outcomes. Clin J Am Soc Nephrol 8: 434-442.
- Stephen C, Noble J, Simpson K, Allison MEM (2006) Long-term quality of life and hospital mortality in patients treated with intermittent or continuous hemodialysis for acute renal and respiratory failure. Ren Fail 28: 323-330.
- Balogun SA, Balogun R, Philbrick J, Abdel-Rahman E (2017) Quality of life, perceptions, and health satisfaction of older adults with end-stage renal disease: A systematic review. J Am Geriatr Soc 65: 777-785.
- Eckardt K-U, Coresh J, Devuyst O, Johnson RJ, Köttgen A, et al. (2013) Evolving importance of kidney disease: from subspecialty to global health burden. Lancet 382: 158-169.
- Cazorla-Vázquez S, Engel FB (2018) Adhesion GPCRs in kidney development and disease. Front Cell Dev Biol 6: 9.
- Pickkers P, Ostermann M, Joannidis M, Zarbock A, Hoste E, et al. (2017) The intensive care medicine agenda on acute kidney injury. Intensive Care Med 43: 1198-1209.
- Muralidharan A, White S (2015) The need for kidney transplantation in low- and middle-income countries in 2012. Transplantation 99: 476-481.
- Luyckx VA, Tonelli M, Stanifer JW (2018) The global burden of kidney disease and the sustainable development goals. Bull World Health Organ 96: 414-422.
- Norton JM, Moxey-Mims MM, Eggers PW, Narva AS, Star RA, et al. (2016) Social determinants of racial disparities in CKD. J Am Soc Nephrol 27: 2576-2595.
- Ojo A (2014) Addressing the global burden of chronic kidney disease through clinical and translational research. Trans Am Clin Climatol Assoc 125: 229-246.
- Sanchez-Niño MD, Sanz AB, Ramos AM, Ruiz-Ortega M, Ortiz A (2017) Translational science in chronic kidney disease. Clin Sci 131: 1617-1629.
- Kam-Tao Li P, Burdmann EA, Mehta RL (2013) Acute kidney injury: global health alert. J Nephropathol 2: 90-97.
- Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, et al. (2012) Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the global burden of disease study 2010. Lancet 380: 2095-2128.
- Humphreys BD (2018) Mechanisms of renal fibrosis. Annu Rev Physiol 80: 309-326.
- Liu Y (2011) Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol 7: 684-696.
- Rao C, Adair T, Bain C, Doi SAR (2012) Mortality from diabetic renal disease: A hidden epidemic. Eur J Public Health 22: 280-284.
- Pickering JW, Endre ZH (2016) Bench to bedside: The next steps for biomarkers in acute kidney injury. Am J Physiol - Ren Physiol 311: F717-F721.
- Alge JL, Arthur JM (2015) Biomarkers of AKI: A review of mechanistic relevance and potential therapeutic implications. Clin J Am Soc Nephrol 10: 147-155.
- Koyner JL, Garg AX, Coca SG, Sint K, Thiessen-Philbrook H, et al. (2012) Biomarkers predict progression of acute kidney injury after cardiac surgery. J Am Soc Nephrol 23: 905-914.
- Meersch M, Volmering S, Zarbock A (2017) Prevention of acute kidney injury. Best Pract Res Clin Anaesthesiol 31: 361-370.
- Schrezenmeier EV, Barasch J, Budde K, Westhoff T, Schmidt-Ott KM (2017) Biomarkers in acute kidney injury - pathophysiological basis and clinical performance. Acta Physiol 219: 556-574.
- Block CA, Schoolwerth AC (2006) Critical care issues for the nephrologist: The epidemiology and outcome of acute renal failure and the impact on chronic kidney disease. Semin Dial 19: 450-454.
- Devarajan P (2008) The future of pediatric acute kidney injury management-biomarkers. Semin Nephrol 28: 493-498.
- Hillege HL, Nitsch D, Pfeffer MA, Swedberg K, McMurray JJV, et al. (2006) Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation 113: 671-678.
- Ichimura T, Hung CC, Yang SA, Stevens JL, Bonventre JV (2004) Kidney injury molecule-1: a tissue and urinary biomarker for nephrotoxicant-induced renal injury. Am J Physiol Physiol 286: F552-F563.
- Medić B, Rovčanin B, Basta Jovanović G, Radojević-Škodrić S, Prostran M (2015) Kidney injury molecule-1 and cardiovascular diseases: From basic science to clinical practice. Biomed Res Int 2015: 1-10.
- Miao J, Friedman E, Wu AHB, Todd JA, Estis J, et al. (2017) Clinical utility of single molecule counting technology for quantification of KIM-1 in patients with heart failure and chronic kidney disease. Clin Biochem 50: 889-895.
- Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, et al. (1998) Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem 273: 4135-4142.
- Campos MAA, Almeida LA de, Grossi MF, Tagliati CA (2018) In vitro evaluation of biomarkers of nephrotoxicity through gene expression using gentamicin. J Biochem Mol Toxicol 32: e22189.
- Silva SCT, de Almeida LA, Soares S, Grossi MF, Valente AMS, et al. (2017) In vitro study of putative genomic biomarkers of nephrotoxicity through differential gene expression using gentamicin. Toxicol Mech Methods 27: 435-441.
- Grossi MF, Campos MAA, Soares S, Silva SCT, Nunes MS, et al. (2017) In vitro study of potential nephrotoxicity biomarkers through gene expression using Amphotericin B. J Toxicol Pharmacol 1: 1008.
- Camacho P, Fan H, Liu Z, He JQ (2016) Small mammalian animal models of heart disease. Am J Cardiovasc Dis 3: 70-80.
- Nath KA. 2015. Models of human AKI: Resemblance, reproducibility, and return on investment. J Am Soc Nephrol 26: 2891-2893.
- Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, et al. (2017) Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrol 28: 218-229.
- Ortiz A, Sanchez-Niño MD, Izquierdo MC, Martin-Cleary C, Garcia-Bermejo L, et al. (2015) Translational value of animal models of kidney failure. Eur J Pharmacol 759: 205-220.
- Bao Y-W, Yuan Y, Chen J-H, Lin W-Q (2018) Kidney disease models: tools to identify mechanisms and potential therapeutic targets. Zool Res 39: 72-86.
- Hayward RS, Harding J, Molloy R, Land L, Longcroft-Neal K, et al. (2018) Adverse effects of a single dose of gentamicin in adults: a systematic review. Bri J Clin Pharmacol 84: 223-238.
- Lavergne A, Vigneau C, Polard E, Triquet L, Rioux-Leclercq N, et al. (2018) Acute kidney injury during treatment with high-dose cloxacillin: a report of 23 cases and literature review. Int J Antimicrob Agents 52: 344-349.
- Quintanilha JCF, Saavedra KF, Visacri MB, Moriel P, Salazar LA (2019) Role of epigenetic mechanisms in cisplatin-induced toxicity. Crit Rev Oncol Hematol 137: 131-142.
- Uccelli A, Moretta L, Pistoia V (2008) Mesenchymal stem cells in health and disease. Nat Rev Immunol 8: 726-736.
- Vanherweghem J-L, Tielemans C, Abramowicz D, Depierreux M, Vanhaelen-Fastre R, et al. (1993) Rapidly progressive interstitial renal fibrosis in young women: association with slimming regimen including Chinese herbs. Lancet 341: 387-391.
- http://wayback.archiveit.org/7993/20171115154508/https://www.fda.gov/Food/RecallsOutbreaksEmergencies/SafetyAlertsAdvisories/ucm096388.htm
- Fink M, Henry M, Tange JD (1987) Experimental folic acid nephropathy. Pathology 19: 143-149.
- Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, et al. (2014) Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci 111: 16836-16841.
- Linkermann A, Bräsen JH, Himmerkus N, Liu S, Huber TB, et al. (2012) Rip1 (Receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int 81: 751-761.
- Khan KM, Jialal I (2018) Folic Acid (Folate) Deficiency. StatPearls 2018: 1.
- Brodsky SV, Satoskar A, Chen J, Nadasdy G, Eagen JW, et al. (2009) Acute kidney injury during warfarin therapy associated with obstructive tubular red blood cell casts: A report of 9 Cases. Am J Kidney Dis 54: 1121-1126.
- Brodsky SV, Nadasdy T, Rovin BH, Satoskar AA, Nadasdy GM, et al. (2011) Warfarin-related nephropathy occurs in patients with and without chronic kidney disease and is associated with an increased mortality rate. Kidney Int 80: 181-189.
- Ware K, Brodsky P, Satoskar AA, Nadasdy T, Nadasdy G, et al. (2011) Warfarin-related nephropathy modeled by nephron reduction and excessive anticoagulation. J Am Soc Nephrol 22: 1856-1862.
- Elterman J, Zonies D, Stewart I, Fang R, Schreiber M (2015) Rhabdomyolysis and acute kidney injury in the injured war fighter. J Trauma Acute Care Surg 79: S171-S174.
- Zhang L, Kang Y, Fu P, Cao Y, Shi Y, et al. (2012) Myoglobin clearance by continuous venous-venous haemofiltration in rhabdomyolysis with acute kidney injury: A case series. Injury 43: 619-623.
- Geng X, Wang Y, Hong Q, Yang J, Zheng W, et al. (2015) Differences in gene expression profiles and signaling pathways in rhabdomyolysis-induced acute kidney injury. Int J Clin Exp Pathol 8: 14087-14098.
- Kim JH, Lee D-W, Jung MH, Cho H-S, Jeon D-H, et al. (2014) Macrophage depletion ameliorates glycerol-induced acute kidney injury in mice. Nephron Exp Nephrol 128: 21-29.
- Singh P, Ricksten SE, Bragadottir G, Redfors B, Nordquist L (2013) Renal oxygenation and haemodynamics in acute kidney injury and chronic kidney disease. Clin Exp Pharmacol Physiol 40: 138-147.
- Tampe B, Steinle U, Tampe D, Carstens JL, Korsten P, et al. (2017) Low-dose hydralazine prevents fibrosis in a murine model of acute kidney injury–to–chronic kidney disease progression. Kidney Int 91: 157-176.
- Endre ZH, Pickering JW (2013) Biomarkers and creatinine in AKI: The trough of disillusionment or the slope of enlightenment? Kidney Int 84: 644-647.
- Ergür BU, Mıcılı SÇ, Yılmaz O, Akokay P (2015) The effects of α-lipoic acid on aortic injury and hypertension in the rat remnant kidney (5/6 nephrectomy) model. Anatol J Cardiol 15: 443-449.
- Tapia E, Soto V, Ortiz-Vega KM, Zarco-Márquez G, Molina-Jijón E, et al. (2012) Curcumin induces nrf2 nuclear translocation and prevents glomerular hypertension, hyperfiltration, oxidant stress, and the decrease in antioxidant enzymes in 5/6 nephrectomized rats. Oxid Med Cell Longev 2012: 1-14.
- Deb DK, Sun T, Wong KE, Zhang Z, Ning G, et al. (2010) Combined vitamin D analog and AT1 receptor antagonist synergistically block the development of kidney disease in a model of type 2 diabetes. Kidney Int 77: 1000-1009.
- Kitada M, Ogura Y, Koya D (2016) Rodent models of diabetic nephropathy: Their utility and limitations. Int J Nephrol Renovasc Dis 9: 279-290.
- Kong L, Wu H, Cui W, Zhou W, Luo P, et al. (2013) Advances in murine models of diabetic nephropathy. J Diabetes Res 2013: 1-10.
- Nishimura A, Kasai T, Kikuno S, Nagasawa K, Okubo M, et al. (2018) Effect of sleep-disordered breathing on albuminuria in 273 patients with Type 2 Diabetes. J Clin Sleep Med 14: 401-407.
- Ruiz-Ortega M, Esteban V, Rupérez M, Sánchez-López E, Rodríguez-Vita J, et al. (2006) Renal and vascular hypertension-induced inflammation: role of angiotensin II. Curr Opin Nephrol Hypertens 15: 159-166.
- Fogo AB (2015) Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol 11: 76-87.
- Daehn I, Casalena G, Zhang T, Shi S, Fenninger F, et al. (2014) Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest 124: 1608-1621.
- Joyce E, Glasner P, Ranganathan S, Swiatecka-Urban A (2017) Tubulointerstitial nephritis: diagnosis, treatment, and monitoring. Pediatr Nephrol 32: 577-587.
- O’Kell AL, Grant DC, Khan SR (2017) Pathogenesis of calcium oxalate urinary stone disease: species comparison of humans, dogs, and cats. Urolithiasis 45: 329-336.
- Fonseca KL, Rodrigues PNS, Olsson IAS, Saraiva M (2017) Experimental study of tuberculosis: From animal models to complex cell systems and organoids. PLoS Pathogens 40: e1006421.
- Eisenbrand G, Pool-Zobel B, Baker V, Balls M, Blaauboer B, et al. (2002) Methods of in vitro toxicology. Food Chem Toxicol 40: 19-236.
- Descotes J (2003) From clinical to human toxicology: linking animal research and risk assessment in man. Toxicol Lett 140–141: 3–10.
- Bernauer U, Oberemm A, Madle S, Gundert-Remy U (2005) The Use of in vitro data in risk assessment. Basic Clin Pharmacol & Toxicol 96: 176-181.
- Innovate UK (2015) A non-animal technologies roadmap for the UK: Advancing predictive biology.
- Morizane R, Bonventre JV (2017) Kidney organoids: A translational journey. Trends Mol Med 23: 246-263.
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, et al. (2014) A comparative encyclopedia of DNA elements in the mouse genome. Nature 515: 355-364.
- Cheng Y, Ma Z, Kim BH, Wu W, Cayting P, et al. (2014) Principles of regulatory information conservation between mouse and human. Nature 515: 371-375.
- Dakhore S, Nayer B, Hasegawa K (2018) Human pluripotent stem cell culture: Current status, challenges, and advancement. Stem Cells Int 2018: 1-17.
- Devuyst O, Knoers NVAM, Remuzzi G, Schaefer F (2014) Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet 383: 1844-1859.
- Stergachis AB, Neph S, Sandstrom R, Haugen E, Reynolds AP, et al. (2014) Conservation of trans-acting circuitry during mammalian regulatory evolution. Nature 515: 365-370.
- PhRMA (2013) 2013 Biopharmaceutical research industry profile. Biopharm Res Ind Phrma 1: 78.
- Marx D, Metzger J, Pejchinovski M, Gil RB, Frantzi M, et al. (2018) Proteomics and metabolomics for AKI diagnosis. Semin Nephrol 38: 63-87.
- Levy SE, Myers RM (2016) Advancements in next-generation sequencing. Annu Rev Genomics Hum Genet 17: 95-115.
- Ouellette AJ, Malt RA, Sukhatme VP, Bonventre JV (1990) Expression of two “immediate early” genes, Egr-1 and c-fos, in response to renal ischemia and during compensatory renal hypertrophy in mice. J Clin Invest 85: 766-771.
- Witzgall R, Brown D, Schwarz C, Bonventre JV (1994) Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J Clin Invest 93: 2175-2188.
- Supavekin S, Zhang W, Kucherlapati R, Kaskel FJ, Moore LC, et al. (2003) Differential gene expression following early renal ischemia/reperfusion. Kidney Int 63: 1714-1724.
- Yuen PST, Jo SK, Holly MK, Hu X, Star RA (2006) Ischemic and nephrotoxic acute renal failure are distinguished by their broad transcriptomic responses. Physiol Genomics 25: 375-386.
- Adler M, Ramm S, Hafner M, Muhlich JL, Gottwald EM, et al. (2016) A quantitative approach to screen for nephrotoxic compounds in vitro. J Am Soc Nephrol 27: 1015-1028.
- Rosner MH, Okusa MD (2006) Acute kidney injury associated with cardiac surgery. Clin J Am Soc Nephrol 1: 19-32.
- Di Carlo C, Brandi J, Cecconi D (2018) Pancreatic cancer stem cells: Perspectives on potential therapeutic approaches of pancreatic ductal adenocarcinoma. World J Stem Cells 10: 172-182.
- Mulligan JK, Patel K, Williamson T, Reaves N, Carroll W, et al. (2018) C3a receptor antagonism as a novel therapeutic target for chronic rhinosinusitis. Mucosal Immunol 11: 1375-1385.
- Neufeld EB, Zadrozny LM, Phillips D, Aponte A, Yu ZX, et al. (2014) Decorin and biglycan retain LDL in disease-prone valvular and aortic subendothelial intimal matrix. Atherosclerosis 233: 113-121.
- Patti GJ, Yanes O, Siuzdak G (2012) Metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol 13: 263-269.
- Salvadori M, Tsalouchos A (2017) Biomarkers in renal transplantation: An updated review. World J Transplant 7: 161.
- Van JAD, Scholey JW, Konvalinka A (2017) Insights into diabetic kidney disease using urinary proteomics and bioinformatics. J Am Soc Nephrol 28: 1050-1061.
- Alonso A, Marsal S, Julià A (2015) Analytical methods in untargeted metabolomics: State of the art in 2015. Front Bioeng Biotech 3: 23.
- Dieterle F, Ross A, Schlotterbeck G, Senn H (2006) Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal Chem 78: 4281-4290.
- Nicholson JK, Lindon JC (2008) Metabonomics. Nature 455: 1054-1056.
- German JB, Hammock BD, Watkins SM (2005) Metabolomics: building on a century of biochemistry to guide human health. Metabolomics 1: 3-9.
- Kell DB, Goodacre R (2014) Metabolomics and systems pharmacology: why and how to model the human metabolic network for drug discovery. Drug Discov Today 19: 171-182.
- Robertson DG, Frevert U (2013) Metabolomics in drug discovery and development. Clin Pharmacol Ther 94: 559-561.
- Sun J, Shannon M, Ando Y, Schnackenberg LK, Khan NA, et al. (2012) Serum metabolomic profiles from patients with acute kidney injury: A pilot study. J Chromatogr B 893–894: 107-113.
- Lin J, Musunuru K (2016) Genome engineering tools for building cellular models of disease. FEBS 283: 3222-3231.
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, et al. (2014) Genome-Scale CRISPR-Cas9 knockout screening in human cells. Science 343: 84-87.
- Shen H, McHale CM, Smith MT, Zhang L (2015) Functional genomic screening approaches in mechanistic toxicology and potential future applications of CRISPR-Cas9. Mutat Res Mutat Res 764: 31-42.
- Hurtado Del Pozo C, Garreta E, Izpisúa Belmonte JC, Montserrat N (2018) Modeling epigenetic modifications in renal development and disease with organoids and genome editing. Dis Model Mech 11: dmm035048.
- Holtzman S (2006) In silico toxicology. Ann N Y Acad Sci 919: 68-74.
- Cronin MTD, Yoon M (2018) Historical context of computational methods to predict toxicity including (Q)SAR, chemoinformatics, grouping, read-across and PBPK modelling. In: Balls M, Combes R, Worth AP, eds. The History of Alternative Test Methods in Toxicology. In press, Academic Press.
- Pletz J, Enoch SJ, Jais DM, Mellor CL, Pawar G, et al. (2018) A critical review of adverse effects to the kidney: mechanisms, data sources, and in silico tools to assist prediction. Expert Opin Drug Metab Toxicol 14: 1225-1253.
- Mostrag A, Yang C, Cronin M, Madden J, Schwab C, et al. (2018) Supporting data location, read-across, and toxicity data gap filling using the COSMOS database. Toxicol 162: 374-375.
- Bitsch A, Jacobi S, Melber C, Wahnschaffe U, Simetska N, et al. (2006) REPDOSE: A database on repeated dose toxicity studies of commercial chemicals—A multifunctional tool. Regul Toxicol Pharmacol 46: 202-210.
- Sakuratani Y, Zhang HQ, Nishikawa S, Yamazaki K, Yamada T, et al. (2013) Hazard Evaluation Support System (HESS) for predicting repeated dose toxicity using toxicological categories. SAR QSAR Environ Res 24: 351-363.
- Richard AM, Judson RS, Houck KA, Grulke CM, Volarath P, et al. (2016) ToxCast Chemical Landscape: Paving the road to 21st century toxicology. Chem Res Toxicol 29: 1225-1251.
- Williams AJ, Grulke CM, Edwards J, McEachran AD, Mansouri K, et al. (2017) The CompTox Chemistry Dashboard: a community data resource for environmental chemistry. J Cheminform 9: 61.
- Patlewicz G, Cronin MTD, Helman G, Lambert JC, Lizarraga LE, et al. (2018) Navigating through the minefield of read-across frameworks: A commentary perspective. Comput Toxicol 6: 39-54.
- Spielmann H, Sauer UG, Mekenyan O (2011) A critical evaluation of the 2011 ECHA reports on compliance with the REACH and CLP regulations and on the use of alternatives to testing on animals for compliance with the REACH regulation. ATLA 39: 481-493.
- Cronin MTD, Richarz A-N (2017) Relationship between adverse outcome pathways and chemistry-based in silico models to predict toxicity. Appl Vitr Toxicol 3: 286-297.
- Schultz TW, Cronin MTD (2017) Lessons learned from read-across case studies for repeated-dose toxicity. Regul Toxicol Pharmacol 88: 185-191.
- Myshkin E, Brennan R, Khasanova T, Sitnik T, Serebriyskaya T, et al. (2012) Prediction of organ toxicity endpoints by qsar modeling based on precise chemical-histopathology annotations. Chem Biol Drug Des 80: 406-416.
- Lei T, Sun H, Kang Y, Zhu F, Liu H, et al. (2017) ADMET evaluation in drug discovery. 18. Reliable prediction of chemical-induced urinary tract toxicity by boosting machine learning approaches. Mol Pharm 14: 3935-3953.
- Su R, Xiong S, Zink D, Loo LH (2016) High-throughput imaging-based nephrotoxicity prediction for xenobiotics with diverse chemical structures. Arch Toxicol 90: 2793-2808.
- Soo JY-C, Jansen J, Masereeuw R, Little MH (2018) Advances in predictive in vitro models of drug-induced nephrotoxicity. Nat Rev Nephrol 14: 378-393.
- Lee K-H, Tseng W-C, Yang C-Y, Tarng D-C (2019) The Anti-inflammatory, anti-oxidative, and anti-apoptotic benefits of stem cells in acute ischemic kidney injury. Int J Mol Sci 20: 3529.
- Li J-S, Li B (2019) Renal injury repair: How about the role of stem cells. In: Liu B, Lan H, Lv L, eds. Renal fibrosis: Mechanisms and therapies. Singapore: Springer. p. 661-670.
- Kolesky DB, Homan KA, Skylar-Scott MA, Lewis JA (2016) Three-dimensional bioprinting of thick vascularized tissues. Proc Natl Acad Sci 113: 3179-3184.
- Kolesky DB, Truby RL, Gladman AS, Busbee TA, Homan KA, et al. (2014) 3D bioprinting of vascularized, heterogeneous cell-laden tissue constructs. Adv Mater 26: 3124-3130.
- Ankley GT, Bennett RS, Erickson RJ, Hoff DJ, Hornung MW, et al. (2010) Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem 29: 730-741.
- Fay KA, Villeneuve DL, LaLone CA, Song Y, Tollefsen KE, et al. (2017) Practical approaches to adverse outcome pathway development and weight-of-evidence evaluation as illustrated by ecotoxicological case studies. Environ Toxicol Chem 36: 1429-1449.
- National Research Council (2007) Toxicity testing in the 21st century. Toxicity testing in the 21st century: A vision and a strategy. Washington DC: National Academies Press pp: 1-196.
- Ferreira AF, França FD, Rossoni JV, Viana PHL, Moraes KCM, et al. (2016) Cyclic adenosine monophosphate protects renal cell lines against amphotericin B toxicity in a PKA-independent manner. Drug Chem Toxicol 39: 28-34.
- França FD, Ferreira AF, Lara RC, Rossoni JV, Costa DC, et al. (2014) Role of protein kinase A signaling pathway in cyclosporine nephrotoxicity. Toxicol Mech Methods 24: 369-376.
- França FD, Ferreira AF, Lara RC, Rossoni JV, Costa DC, et al. (2014) Alteration in cellular viability, pro-inflammatory cytokines and nitric oxide production in nephrotoxicity generation by Amphotericin B: involvement of PKA pathway signaling. J Appl Toxicol 34: 1285-1292.
- Nelms MD, Mellor CL, Cronin MTD, Madden JC, Enoch SJ (2015) Development of an in silico profiler for mitochondrial toxicity. Chem Res Toxicol 28: 1891-1902.