Dilated cardiomyopathy (DCM), characterized by dilatation of cardiac chambers and systolic dysfunction, is one of the leading causes of severe heart failure (HF) and the most common indication for heart transplantation. Despite significant therapeutic improvements, its incidence and mortality is persistently high largely attributable to heterogeneous aetiology. Infectious diseases and genetic factors predominate research on the causes of DCM. However, evidence over the past two decades reveal distinct autoantibodies or other immune factors in heterogeneous subsets of DCM suggesting the involvement of autoimmune mechanisms in the pathogenesis of DCM. Hitherto, accumulated evidence demonstrates abnormalities in cell-mediated immunity such as altered lymphocyte function and relative proportion of lymphocytes subsets, activated cytokine system, inappropriate expression of histocompatibility complex on cardiac tissue and the expression of adhesion molecules on cardiomyocytes. This evidence suggest a subset of DCM patients is autoimmunity-mediated. This review aims to summarize the contemporary understanding of the characteristics of autoimmune cardiomyopathy in humans, proposed pathophysiology mechanisms as well as the ongoing clinical investigations that have focused on autoimmunity as a target for therapy.
autoimmune cardiomyopathy, autoimmune heart failure, inflammatory cardiomyopathy
Abbreviations
AAB: Autoantibodies; AAG: Autoantigens; ACCF: American College of Cardiology Foundation;
ACE-I: Angiotensin-Converting-Enzyme Inhibitor; AD: Autoimmune Diseases; AHA: American Heart Associations; ARBs: Angiotensin Receptor II Blockers; AS: Ankylosing Spondylitis; AV: Atrioventricular; CAD: Coronary Artery Disease; CI: Confidence Intervals; CM: Cardiomyopathy; DCM: Dilated Cardiomyopathy; ECG: Electrocardiogram; Echo: Echocardiography; EMB: Endomyocardial Biopsy; EMB: Endomyocardial Biopsy; ESC: European Society of Cardiology; ESR: Erythrocyte Sedimentation Rate; EULAR: European League against Rheumatism; EUSTAR: European League against Rheumatism Scleroderma Trials and Research; GCM: Giant Cell Myocarditis; GPA: Granulomatosis with Polyangiitis; HBV: Hepatitis B Virus; HF: Heart Failure; HLA: Human Leukocyte Antigen; iDCM: Idiopathic Dilated Cardiomyopathy; IVIG: Intravenous Immunoglobulin; LGE: Late Gadolinium Enhancement; LV: Left Ventricular; LVEF: Left Ventricular Ejection Fraction; MC: Myocarditis; MI: Myocardial Infarction; MRI: Magnetic Resonance Imaging; NSAID: Non-Steroidal Anti-Inflammatory Drugs; PAN: Periarteritis Nodosa; RA: Rheumatoid Arthritis; RV: Right Ventricular; SCD: Sudden Cardiac Death; SLE: Systemic Lupus Erythematosus; TNF-α: Tumour Necrosis Factor-Alpha; VT: Ventricular Tachycardia
The human immune system is an intricate set of cellular, chemical and soluble protein mechanisms that shield the body against alien substances such as infections and tumour cells without attacking self-molecules [1]. However, in the setting of an autoimmune disease (AD), the immune system initiates an attack on self-molecules due to the deterioration of immunological tolerance to autoreactive immune cells [2,3]. Although ADs are relatively uncommon with an overall prevalence of 3% to 5% in the general population, their effect on mortality and morbidity are clinically significant [4,5]. Genetics, infection and/or environmental factors may play an active role in the initiation of autoimmune attacks against the body’s self-molecules [6]. ADs are an important aetiology of dilated cardiomyopathy (DCM) secondary to myocarditis (MC). DCM is defined as the dilatation of cardiac chambers leading to the impairment of systolic function [7,8] and heart failure (HF) for which the only definitive treatment is cardiac transplantation because fibre damage and fibrosis are irreversible processes [9]. Other prevalent aetiologies of DCM due to inflammation and autoimmune mechanisms are infectious diseases and chronic exposure to environmental toxins and/or cytotoxic drugs [10]. The present review focuses on ADs as one of the leading aetiologies of CM, discusses their involvement in the pathophysiology of CM and concludes with a meta-analysis of diagnostic and clinical management strategies.
Autoimmune diseases: Typically, ADs develop in the setting of a defective function of any features contributing to the non-reactivity of the immune system towards self-antigens characterized by high concentrations of autoantibodies (AAB) reacting with autoantigens (AAG) [9,11,12]. High concentrations of AABs and AAGs play a central role in the diagnosis and disease classification as well as are involved in tissue damage [12]. Definitionally, AABs are markers of immune response resulting from a variety of stimuli including infections, drugs, environmental agents, food and metals, or prior lesions secondary to tissue destruction while AAGs are molecules capable of reacting with effector arms of a patient’s own immune response [13-17]. AABs target a wide spectrum of AAGs located in the organ containing the autoimmune lesion. The pathogenesis of AABs depend on its activity, accessibility of antigenic targets and actions of the immune response effector mechanisms [18]. Although various diseases with cardiologic effects such as rheumatic fever, myocarditis (MC), DCM, post-pericardiotomy syndromes and collagen disease may exhibit immunopathological responses, they do not fall under the typical definition of ADs [18]. In patients with CM, a great variety of AABs that attack cardiac AAGs may be pathogenic agents or epiphenomena of tissue aggression and their presence in biological samples represents an important diagnostic marker for illness with pathological significance if accompanied by an illness caused by an autoimmune response [18-22]. Connective tissues, granulomatous and eosinophilic diseases are among the most common ADs capable of causing CM [10,23].
Connective tissue diseases: Definitionally, connective tissue diseases are disorders characterized by disruptions in the cells and matrix that bind together body tissues. These diseases frequently involve the joints, muscles and skin, as well as organs and organ system such as the heart, lungs, kidney, gastrointestinal (GI) tract, blood vessels and eyes [24]. Although more than 200 diseases can affect connective tissues, only six have been extensively described as frequent aetiologies of CM. These are systemic lupus erythematosus (SLE), rheumatoid arthritis, ankylosing spondylitis, scleroderma, dermatomyositis and periarteritis [23].
Systemic lupus erythematosus: Systemic lupus erythematosus (SLE) is a chronic inflammatory disease characterized by an AABs reaction against nuclear and cytoplasmic antigens. The disease is more prevalent (> 90%) in women, frequently beginning to manifest at childbearing age [25,26]. SLE has a protean presentation with a relapsing and remitting course. It can affect almost any organ system although it mainly involves the skin, joints, kidneys, blood cells, nervous system and the heart [25]. Cardiovascular involvement might be rare but remains a long-term concern even after 4 to 20 years since the initial diagnosis [23]. Despite improvement in therapy, cardiovascular mortality remains significant for SLE patients [27,28]. A number of cardiac abnormalities may manifest in SLE patients but the development of DCM is not a prominent manifestation of the natural history of the disease. Global left ventricular (LV) dysfunction, segmental LV wall motion abnormalities and right ventricular (RV) enlargement affect 5%, 4% and 4% of SLE patients respectively, and these complications correlate with disease severity [29].
Patients with SLE are three-fold more likely to be hospitalized with newly diagnosed HF compared to healthy controls with the greatest risk in women younger than 45 years of age [30]. SLE induces CM and HF through atherosclerosis, inflammation, and drug-induced impairments [23]. SLE patients have 2- to 10-fold higher risk for myocardial infarction (MI) and progressive atherosclerosis compared to healthy age-matched controls. Atherosclerosis is the primary cause of HF in 20% of SLE patients [31]. Co-occurrence of antiphospholipid antibodies increases the risk of thrombosis-mediated MI and HF. Diagnosis of myocardial inflammation is frequent at autopsy or at endomyocardial biopsy (EMB) in greater than 50% of the patients compared to clinical detection, which is about 9% [32-34]. Clinical presentation of SLE-associated MC is often undistinguishable from other forms of MC. Treatment for SLE using hydroxychloroquine may cause CM with variable impact on mortality [35] and associated pathological findings such as progressive bundle branch block and atrioventricular (AV) heart block and biventricular hypertrophy [36,37].
Diagnosis of SLE-mediated HF includes a combination of electrocardiograph (ECG), standard echocardiography and coronary angiography. These tests do not diagnose SLE as the primary aetiology and thus may require EMB to delineate SLE as the primary cause. However, findings of EMB are often non-specific with myocardial lesions characterized by increased interstitial connective tissue and myocardial scarring [38]. Depolarization abnormalities on signal-averaged ECG accompanied by echocardiographic results of abnormal LV filling may suggest the presence of myocardial fibrosis and can be a marker of subclinical myocardial involvement in SLE patients [38]. Cardiac involvement manifests as conduction system abnormalities such as complete AV heart block. Cardiac magnetic resonance imaging (MRI) and myocardial stain or stain rate can identify early manifestations of SLE-induced CM [39,40].
The American College of Cardiology Foundation/American Heart Association (ACCF/AHA) recommends that treatment for SLE-induced CM and HF should be dependent on the underlying cause mainly myocarditis (inflammation), thrombosis or atherosclerosis. Treatment for SLE-associated MC should be intravenous methylprednisolone followed by a high dose oral prednisone for 1-2 weeks [41]. Small studies have also reported the use of intravenous immunoglobulin, azathioprine and cyclophosphamide for SLE-related MC [42-44]. Treatment for thrombosis and atherosclerosis should be standard percutaneous and pharmacological interventions as detailed in other guidelines for acute MI [45], and chronic CAD and long-standing HF as detailed in the European Society of Cardiology (ESC) HF guidelines [46]. Common comorbid diseases such as hypertension should also be treated [23].
Rheumatoid arthritis: Rheumatoid arthritis (RA) refers to a chronic progressive and disabling autoimmune disease. The disease causes inflammation, swelling, and pain in joints and other body organs [47]. Cardiac involvement in RA results from the development of MC or pericarditis while HF may result from RA medication, MC, vasculitis, valvular involvement, progressive atherosclerosis or MI, and amyloidosis but the development of CM is rare in RA patients with HF [48]. A retrospective study of 172 patients reveals that symptomatic cardiac involvement, which includes MC, perimyocarditis and pericarditis, affects 7.6% of RA patients [49]. The presence of RA increases the risk of HF two-fold compared to non-RA patients followed for a prolonged period of 46 years with a significantly greater risk for women [50]. The presence of MC and pericarditis are poor prognostic markers for RA patients [23].
Pathogenic mechanism of myocardial involvement in RA is not well documented, although it may result from disturbances in the microcirculation with microvasculitis in the absence of ECG abnormalities. Although chronic inflammation may contribute to the pathogenesis of CM, the mechanisms remain unclear. Elevated serum biomarkers such as C-reactive protein, interleukin-6 and tumour necrosis factor-alpha suggest the development of HF and mortality in a subset of RA patients already diagnosed with HF [51]. In patients with RA and HF, the presence of elevated erythrocyte sedimentation rate (ESR) levels, RA vasculitis or RA lung disease is an independent predictor of worse mortality [23].
In addition to specific data on the diagnosis of RA-associated CM lacking, RA-mediated HF lacks pathognomonic diagnostic features and diagnosis rests on strong clinical suspicion. Tests for serum levels of biomarkers (mainly B-type natriuretic peptides [BNP]), echocardiography and angiography to exclude atherosclerosis as an intermediate cause may support the diagnosis of RA. Cardiac injury in RA patients may present as pericardial effusion, diastolic dysfunction and evidence of inflammation on cardiac MRI and nuclear scans [23]. EMB may be useful for revealing a diffuse necrotizing or granulomatous pattern but is non-specific [52]. Treatment for RA-associated CM with signs and symptoms of HF depend on the underlying cause and treatment should be according to current ESC and ACCF/AHA HF guidelines [46,53]. RA patients with asymptomatic LV dysfunction may receive standardized HF medications including angiotensin-converting-enzyme inhibitor (ACE-I) and beta-blockers according to the current updated HF guidelines [46,53].
Periarteritis nodosa: Periarteritis Nodosa (PAN) is an acute or sub-acute medium-size vessel necrotizing vasculitis mainly involving arteries disease affecting patients of any age and prevalent in both genders [54]. It is a rare vasculitis often associated with Hepatitis B virus (HBV). At present, the incidence of PAN is much lower than in the past. In systematic PAN, frequent clinical manifestations often involve peripheral nerves, skin, GI tract, heart and kidneys [55]. Cardiac involvement is rare but severe and often presages poor clinical outcomes. In cardiac involvement, coronary arteries and their branches, and the myocardium are more frequent targets than the pericardium and endocardium [55]. In a series of 348 patients with PAN, the incidence of severe hypertension was 6.9% and CM 7.5% with a higher risk in RA patients with HBV [56]. MC is a rare cause of HF in PAN patients but with an ominous prognosis [23].
The diagnosis of PAN lacks pathognomonic features and depends on a high level of clinical suspicion. In patients with biopsy-proven PAN, there should be a high clinical suspicion with progressive angina and deterioration of LV function. Angiography may be useful for diagnosis [23]. Early treatment is the best hope for better outcomes [55]. Common treatment strategy include aggressive immunosuppression with steroids and selective revascularization with possible treatment with antiviral agents for co-occurring HBV [23]. Treatment of PAN in the absence of HBV requires a combination of corticosteroids and cyclophosphamide although sequelae of cardiac involvement may occur despite treatment [55].
Scleroderma: Scleroderma refers to an autoimmune disorder affecting the skin and many internal organs in the setting of excessive production of collagen protein, which normally strengthens and supports connective tissues in the body [57]. Scleroderma patients exhibit diffuse vascular damage, abnormal activation of the immune system and fibrosis of the skin and many internal organs [58]. Fibrosis can also affect many internal organs leasing to the impairment or failure of the affected organ. Primary cardiac involvement has long been recognized as a complication of scleroderma (although frequently underdiagnosed) associated with major adverse prognostic impact that can be potentially treatable when diagnosed in time [59]. In a systematic review of 18 cross-sectional and prospective studies describing 4,854 patients, the prevalence of cardiac involvement ranged between 7% and 39% although there was significant heterogeneity in the definition of cardiac disease [60]. Scleroderma portends greater mortality and various cardiac manifestations including HF, arrhythmias and diastolic dysfunction [60].
Limited evidence associates scleroderma-associated CM with poor prognostic indicators including diffuse cutaneous disease, positive scleroderma-specific serology, older age at disease onset, tendon friction runs, abnormal nail fold capillaroscopy, and worse quality of life scores [60]. Echocardiographic study reveals no marked differences in LV dimensions or fractional shortening but may indicate systolic impairment in a majority of patients [61]. The EULAR (the European League against Rheumatism) Scleroderma Trials and Research (EUSTAR) cohort attributed 26% of fatal scleroderma patients to HF [62]. Half of scleroderma patients exhibit a distinctive focal myocardial lesion that ranges from band necrosis to replacement fibrosis in the absence of structural abnormalities of the coronary arteries. Progressive systematic scleroderma can result in conduction abnormalities, arrhythmias, HF, angina pectoris and sudden cardiac death (SCD) [23]. Cardiac MRI with late gadolinium enhancement (LGE) may be useful in characterizing patchy fibrosis and myocardial oedema interspersed with normal myocardium [63]. Cardiac dysfunction in systemic scleroderma portends an ominous prognosis directly related to the degree of myocardial fibrosis [23].
Diagnosis of scleroderma-associated CM lacks a specific definitive test and may require a high index of clinical suspicion. Although non-specific, ECG abnormalities may include AV block and sinus node disease, and Holter monitoring may show ventricular arrhythmias and atrial fibrillation. Speckle tracking echocardiography imaging may be useful in assessing longitudinal strain and demonstrating early changes associated with scleroderma HF while cardiac MRI may reveal patchy fibrosis [64]. Patients with scleroderma with evidence of skeletal myopathy are 2.5 times more likely to develop HF or arrhythmias compared to patients without skeletal myopathy [65]. They also are more likely to have autoantibodies (anti-PM/Scl antibodies and anti-ribonucleoprotein antibody) [65]. Although EMB may be useful in the diagnosis of acute MC when there is clinical suspicion, it should be based on favourable response to intravenous methylprednisolone [66]. Clinical management of scleroderma CM and HF is problematic because of the lack of a specific treatment regimen. However, careful use of vasodilator therapy should be considered because relative hypotension might prevent aggressive up-titration of traditional HF medication. Autologous stem cell transplantation may be useful but usually contraindicated in patients with HF, and routine steroid therapy could aggravate the disease [67]. Treatment of concomitant pulmonary arterial hypertension may relieve symptoms and heart transplantation considered in patients with low systemic burden of scleroderma [68].
Dermatomyositis: Dermatomyositis belongs to idiopathic inflammatory myopathy with characteristic cutaneous findings that affect both children and adults. The disease includes polymyositis and inclusion-body myositis and often affects the joints, oesophagus, the lungs and less commonly the heart [69,70]. Although uncommon, cardiovascular involvement constitute a major cause of death in patients with dermatomyositis. MC maybe silent and possibly under-diagnosed [71]. The most common cardiac manifestations is HF constituting 32-77% of total cardiovascular events in a meta-analysis of 1,530 patients [72]. Based on the findings of autopsy studies, other common cardiac manifestations including MC (25%), focal fibrosis (25%) and skeletal involvement in all cases of HF [73], abnormal ECGs (65%) and MC (30%) [74]. Additionally, LV dysfunction secondary to MC and arrhythmias particularly conduction abnormalities that may lead to third degree AV block whereas syncope are frequent cardiac manifestations. Ventricular ectopic beats are frequent but ventricular tachycardia (VT) is rare [71]. In addition to be a cause of DCM, a case study has associated dermatomyositis with the development of restrictive CM in patients with dermatomyositis [75].
Diagnosis of dermatomyositis is not straightforward. Creatine kinase isoenzyme and cardiac troponin can be useful as markers of overall disease severity but are non-specific since they can be elevated even in the absence of cardiac manifestations of dermatomyositis [76]. In these patients, HF can develop even in the absence of systemic inflammation. DCM could result from direct mononuclear inflammatory cell infiltrates leading to the degeneration of cardiomyocytes or it can be a consequence of progressive atherosclerosis or vasculitis leading to necrosis [77]. As a result, EMB may detect inflammation, necrosis and degenerative changes similar to those observed in skeletal muscles. However, EMB is non-specific and thus infrequently used in clinical practice. ECG abnormalities may be common but are also non-specific [23,71].
Although cardiac manifestations could be potentially fatal in dermatomyositis patients, the prevalence of cardiac disorders and their therapeutic approach is not well established. Based on sporadic reports, corticosteroids and immunosuppressive therapies in patients with untreated dermatomyositis presenting with MC reduces the size of myocardial damage and improves wall hypokinesia [78]. Treatment of patients with dermatomyositis often focuses on standard HF therapy. Intravenous methylprednisolone may cause some improvement in patients with cardiac MRI evidence of early cardiac manifestation of dermatomyositis and heart transplantation can be used only rarely in patients without significant systemic involvement of dermatomyositis [78]. There are no data on therapeutic intervention for patients already under treatment or in disease remission. In these cases, the use of ICD therapy may prove clinically beneficial [71].
Ankylosing spondylitis: Ankylosing spondylitis (AS) describes a chronic, inflammatory and rheumatic disease with a predilection for the sacroiliac joint and spine, and less frequently peripheral joints. AS can also present with non-joint involvement. The disease most often affects young adult males and may lead to spinal vertebrate fusion [79,80]. The human leukocyte antigen (HLA)-B27 is present in the majority of patients with AS believed to play a role in the pathophysiologic manifestations of the disease [81]. Non-joint involvement may present as ophthalmological, pulmonary, neurological and cardiac manifestations. Cardiac manifestations in AS patients is uncommon, occurring in about 2-10% of patients [80,81]. Initially in 1936, aortitis was found in a group of patients with spondylitis [81]. The presence of aortic valve disease in a subgroup of patients with rheumatoid spondylitis was one feature that helped to distinguish rheumatoid arthritis and ankylosing spondylitis as two distinct diseases [80]. Today, it is widely accepted that AS may present not only with aortic pathology but also with conduction defects, valvular regurgitation and CM [82,83]. Recognition of these changes is important since in many patients cardiac changes may begin prior to the onset of clinical symptoms [81]. Cardiac involvement may be in the form of aortic insufficiency, aortitis at the ascending aorta, AV block or branch block. The most frequently observed complications are conduction defects and aortic insufficiency [84,85]. Mitral insufficiency is rare but is a potential cause of cardiac failure [86].
Myocardial dysfunction observed in AS patients is that of the diastolic variety as opposed to systolic. Yildirir et al. [85] found significant diastolic dysfunction in AS patients particularly abnormal relaxation pattern demonstrated on echocardiography by lower E-wave velocity (early diastolic filling), higher A-wave velocity (late diastolic filling), and an overall low E/A ratio. Similarly, Gould et al. [87] investigated peak filling throughout exercise along with time to achieve peak filling during strenuous activity using radionuclide angiography and observed a significant reduction in both measures in AS patients compared to healthy volunteers. Thus, these findings support an overall decrease in compliance suggesting a reduction in diastolic function. Inflammatory infiltrates, cytokines, genetic and environmental factors may contribute to the aetiology of AS. Chronic, systemic and inflammatory states within AS may also cause cardiac and vascular pathologies [88]. Cardiovascular mortality increases in AS patients, with older age, higher ESR levels and more inflamed peripheral joints portends a worse prognosis [84]. The most frequently observed symptom in AS patients is chest pain, which may present with atypical findings such as having no pain or the presence of cardiac, neurological or ischemic extremities [79].
For several decades, the mainstay therapy of AS has been non-steroidal anti-inflammatory drugs (NSAIDs). Although a recent study associated NSAIDs with adverse CVD events [89], another study on inflammatory polyarthritis patients taking NSAID failed to demonstrate an increased rate of CVD deaths [90]. Similarly, a cohort study in Norway reported that increasing the levels of C-reactive proteins, delayed AS diagnosis, work disability and infrequent use of NSAISs could independently predict the mortality of AS patients suggesting that the risk of NSAID treatment might be offset by the benefits of inflammation suppression [91]. Tumour necrosis factor-alpha (TNF-α) antagonists are an alternative for patients non-responsive or intolerant to NSAIDs [92] but should be prescribed with caution to patients with HF because of reports of increased mortality and morbidity [93]. Nevertheless, TNF-α antagonists reduce CVD events partly because they work by suppressing systemic inflammation [94-96].
Granulomatous diseases: Granulomatous diseases comprise a large family sharing histological denominator of granuloma formation. Granuloma are focal compact collection of inflammatory cells in which mononuclear cells predominate (macrophages and their derivatives the epithelioid cells), which are evoked by the persistence of poorly- or non-degradable substances or due to active cell-mediated hypersensitivity responses [97]. An overlap of the two mechanisms may occur in most infectious diseases because microorganisms can serve as both foreign bodies and antigens for immunologic responses [98]. Normally, granulomas result from protective mechanisms and form when acute inflammatory processes cannot destroy invading agents. Various granulomas may contain lymphocytes, sometimes plasma cells, giant cells, eosinophils, mast cells and fibroblasts [97]. Based on causative agents, granulomas have a diverse aetiology classified into four groups: (i) T-cell-mediated immune granulomas formed in response against infectious agents; (ii) granulomas with unknown aetiology but with T lymphocyte-mediated profile; (iii) foreign body granulomas induced by inanimate substances; and (iv) granulomas associated with malignant tumours [97]. Granulomatous diseases well known to cause CM in humans include sarcoidosis, granulomatosis with polyangiitis and granulomatous MC.
Sarcoidosis: Sarcoidosis refers to a multi-system inflammatory disease of unknown aetiology characterized by the presence of T lymphocytes, mononuclear phagocytes and noncaseating granulomas in the affected tissues [99]. Frequently, sarcoidosis involves multiple organ systems including the eyes, skin, lungs, lymphatic system and the heart. The disease is common among the young and middle-aged adults although children and the elderly may also be affected and is more prevalent in women than in men [100]. Sarcoidosis is clearly an inflammatory disease but genetics environmental exposures and infectious diseases may also play a role in its pathogenesis [10]. Cardiac involvement occurs in about 20 to 50% of sarcoidosis patients based on autopsy series but only a small fraction have clinically recognized disease. Following the early stages of sarcoidosis, characterized by granulomas inflammation, the majority of the cases are clinically silent and can resolve or progress to end-organ fibrosis [23]. As sarcoidosis becomes clinically apparent, incidence of HF, conduction defects, and SCD increase [101,102]. Myocardial involvement in patients with sarcoidosis is common although the pericardium and endocardium can also be involved [103]. Typical features of myocardial involvement include conduction defects, VT, aneurysm formation or global systolic dysfunction. About 5% of sarcoidosis patients who present with a predominant cardiac disease but in the absence of the characteristic pulmonary, dermatologic or ocular features [104]. Focus on cardiac disease is important since it is among the leading cause of death in sarcoidosis patients but which is potentially preventable by the use of early and appropriate therapy [23].
Diagnosis of cardiac sarcoidosis lacks a reliable reference standard as well as pathognomonic diagnostic features. Its characteristic pathological lesion (noncaseating granuloma) can also result from various infectious and non-infectious diseases [101,102,104,105]. The AHA guidelines on the diagnosis and treatment for specific types of DCM recommend that diagnosis of sarcoidosis should depend on pathological demonstration of cardiac granulomas or non-invasive evidence of cardiac involvement in a patient with pathologically proven extra-cardiac sarcoidosis [103]. Initial tests should include detailed history, physical examination, ECG and chest radiograph. Common ECG abnormalities in patients with sarcoidosis include bundle branch block, new AV-block, frequent premature ventricular complexes, VT, pathological Q waves and ST-T changes, and a first-degree heart block progressing into a complete heart block – observed in patients with clinically apparent disease. [104,105]. Although ECG abnormalities are non-specific, they should prompt further clinical investigation [103]. The presence of cardiac symptoms, ECG abnormalities and/or cardiomegaly on chest radiograph should prompt also for further non-invasive investigation. [23]. Echocardiography is essential to assess for ventricular systolic and diastolic dysfunction, and abnormalities in wall-motion, wall thickness, and Doppler filling patterns, which are frequent findings that suggest cardiac sarcoidosis. Holter monitoring is important since 100 ventricular ectopic beats within 24 hours is suggestive of sarcoidosis [101]. On cardiac MRI, early enhancement of sarcoid granulomas in T2-weighted gadolinium suggests inflammation and oedema, while late enhancement suggests fibrotic changes and scarring [106-108]. If available, cardiac MRI should be the choice imaging modality for patients with sarcoidosis [106,107].
In the clinical management of cardiac sarcoidosis, corticosteroids is the mainstay therapy for patients useful for suppressing inflammation and granuloma formation. However, there is a paucity of data to support the effectiveness of corticosteroid therapy as well as clear guidelines on when to initiate therapy [100]. Nevertheless, most experts agree that clinicians should consider immunosuppression in symptomatic patients with cardiac sarcoidosis with evidence of active myocardial inflammation and any of the following: (i) reduced LVEF; (ii) high grade AV-block; (iii) frequent premature ventricular contraction or non-sustained VT; or sustained VT or ventricular fibrillation [100]. Based on current evidence, it is uncertain whether asymptomatic patients can benefit from corticosteroid therapy [109]. In sarcoidosis patients with reduced LVEF, guidelines-directed medical therapy for HF, including ACE-I, angiotensin receptor II blockers (ARBs), diuretics, aldosterone inhibitors should be initiated. Beta-blockers should be used with caution in patients without pacemaker due to increased risk of high grave AV-block. Digoxin should be avoided in the acute stage of cardiac sarcoidosis due to the risk of heart block and arrhythmias in the setting of active myocardial inflammation [23,100]. Catheter radiofrequency ablation may be useful in cardiac sarcoidosis patients with VT, and in patients who are non-responsive, catheter ablation may be considered. In patients with advanced HF, device therapy, mechanical circulatory support or heart transplantation may be considered [100].
Granulomatosis with polyangiitis: Granulomatosis with polyangiitis (GPA), formerly known as Wegener's granulomatosis is among the rarest but potentially life-threatening systemic necrotizing vasculitis predominantly affecting small and medium sized vessels [110,111]. The upper and lower respiratory system, systemic vasculitis, and necrotizing glomerulonephritis are the characteristic components of the disease triad [112]. Overtime, the prevalence has increased from 3.0 to 15.7 per 100,000 patients but this increase may simply be a reflection of the availability of new and more accurate diagnostic modalities and serologic tests [112-114]. GPA can manifest in multiple organ systems and presents with a variety of symptoms. Cardiac involvement is rarely documented antemortem with an estimated incidence of 3.3% to 13% [111,115,116]. Besides increasing incidence of GPA, the difference in the incidence may also be due to heterogeneity of cohorts, different definitions of cardiac manifestations and different diagnostic tools used [117].
GPA may involve any portion of the heart with pericarditis, myocarditis and conduction system defects being the most frequent. Manifestations such as pericarditis due to serositis have been described in GPA previously [118-121]. In a study of three GPA patients, Grant et al. associated cardiac complications with inflammation secondary to ANCA positive vasculitis [122]. However, reports of cases of CM with reduced LVEF are infrequent in GPA patients [123]. Necrotizing vasculitis or granulomatous inflammation involving the myocardium or coronary arteritis may give rise to conduction defects, lethal arrhythmias, cardiomyopathies or coronary aneurysms [117]. Ventricular arrhythmias triggered by active vasculitis are uncommon and rarely reported. Conduction abnormalities, valvular disorders, unstable angina secondary to vasculitis and subclinical MC are uncommon cardiac manifestation of GPA [123-125]. DCM and HF in GPA patients is exceedingly exceptional with only eight cases reported in literature since its seminal description by Wegner in 1936 [126-132].
Pathophysiologically, the presence of granulomas and vasculitis suggest an exaggerated cellular immune or hypersensitive response although its aetiology remains unclear. The preponderance of GPA in the respiratory tract suggests inhaled antigens may initiate cellular immune response. T cells, monocytes and neutrophils comprise the key cellular elements to indicate the combined operation of cell-mediated and neutrophil-mediated immune mechanisms [117]. Neutrophils are the likely primary mediators of early myocardial injury while mononuclear phagocytes and lymphocytes involvement occurs in the late phases of vasculitis process. Circulating PR3-ANCAs occur in 70 – 93% of patients with entreated GPA suggesting the role of AABs in the pathogenesis and natural course of GPA [133-136]. The role of genes in influencing the susceptibility to GPA has not been well-described but major histocompatibility complex alleles or genetic polymorphisms may play a role in the pathophysiology of GPA [133,137-139].
Diagnosis of GPA is difficult and often delayed due to a wide range of clinical presentations. Typically, GPA is associated with a positive antineutrophil cytoplasmic antibody (ANCA) but this is not always the case [112]. Fauci et al. [126] proposed diagnostic criteria of GPA based on clinical evidence of disease in at least two of the three areas – upper airways, lung and kidney – and biopsy results showing disease in at least one and preferably two of these organs. Diagnostic criteria established by American College of Rheumatology to distinguish GPA from other vasculitis: (i) urinary sediment containing red blood cells per high-power field; (ii) abnormal findings on the chest radiograph; (iii) oral ulcers or nasal discharge; and (iv) granulomatous inflammation on biopsy [140]. Specific diagnosis of GPA-associated CM rests on patient’s history, laboratory investigation and non-invasive imaging. In a case report, ECG abnormalities included sinus tachycardia with T-wave flattening, cardiomegaly on chest x-ray examination, elevated cardiac troponin; on echocardiography a severe dilated CM with severely reduced LVEF; and on cardiac MRI, multiple foci of delayed enhancement typical of myocardial necrosis and scarring. The presence of a positive ANCA usually confirms the diagnosis of GPA [141].
Prior to the advent of effective therapy, mean survival among patients with untreated active GPA was < 6 months and > 80% died within three years of onset of symptoms [142,143]. Corticosteroids ameliorated many of the inflammatory manifestations of GPA although it only resulted in modest improvement in survival (a mean survival of only 12 months) [144]. Studies in the 1970s investigating a dual therapy of cyclophosphamide and corticosteroids (tapered and discontinued 6-9 months) significantly improved the prognosis of GPA [126,143,145]. The dual therapy reduced 5-year mortality rates to < 15%, relapses noted in 30-70% of patients by re-introduction of therapy is often effective [146,147]. However, sequelae of vasculitis such as cerebrovascular, MI, renal failure and hypertension, or cyclophosphamide-associated complications such as opportunistic infections and neoplasms contributed to the late mortality and morbidity [148-150].
Other inflammatory diseases
Giant cell myocarditis: Giant cell myocarditis (GCM) is a rare cardiac disease characterized by severe myocardial inflammation. Early data on GCM documented in case reports and case series before the advent of heart transplantation and sophisticated immunosuppressive therapy reveal that in a majority of patients with GPA, diagnosis was made at autopsy because of a sudden onset and rapid deterioration of GCM. Typically, GCM affects the young (mean age 40 years) and about 20% have a history of autoimmune disorders [152,153]. A majority of patients present with acute HF and 50% will have refractory VT during the course of GDM. Patients may also present with varying extent of heart block. GCM has an ominous prognosis with median survival of period of approximately three months for patients without heart transplant or immunosuppression [152]. While the aetiology of GCM remains not fully elucidated, current evidence suggests it may arise due to immune dysregulation mediated by T lymphocytes. Rats immunized with cardiac myosin developed GCM [154] and gene analysis of GCM patients reveal increased immune response, in particular an upregulation of chemokines involved in the activation of Th1 cells [155]. Furthermore, GCM patients do not exhibit significant presence of cardiac AABs as observed in lymphocytic MC suggesting an autoimmune response dominated by T cells rather than B cells, which is consistent with a positive therapeutic response to T-cell directed immunosuppression [156].
Since giant cells are the key characteristics feature of GCM, differential diagnosis of GCM includes other myocardial lesions containing giant cells such as infectious diseases, systemic granulomatous processes and foreign body giant cell reaction, foremost among these diseases are cardiac sarcoidosis and fulminant lymphocytic MC [157]. In contrast to GCM, cardiac sarcoidosis is characterized by tight non-necrotizing granulomas that often have central multinucleated giant cells [158,159]. The diagnosis of GCM rests on histological tests and EMB, which is frequently made at post-mortem or from an explanted heart. Typical histological findings include serpiginous necrosis, characteristic multinucleated giant cells, and a mixed inflammatory infiltrate of lymphocytes, histocytes and frequently eosinophils [10]. If clinical suspicion is high despite a negative initial EMB and imaging studies or other clinical findings strongly suggest GCM, a repeat EMB is advisable, which improves diagnostic accuracy from sensitivity from 68% to 93% in one study [153]. EMB is useful in confirming diagnosis of MC and identifies the underlying aetiology and the type of inflammation – giant cell, eosinophilic MC or sarcoidosis [160]. Standard 12-lead ECG should be performed on all patients suspected of having GCM. EGC abnormalities are often abnormal in MC although they are neither specific nor sensitive. AV block accompanied by mild LV dilatation can be due to various causes including laminopathy but it may suggest GCM, cardiac sarcoidosis or Lyme disease [160]. Echocardiography is useful to exclude non-inflammatory cardiac causes such as valvular disease and to monitor changes in cardiac chamber size, wall thickness, ventricular function and pericardial effusions [161].
In the absence of appropriate therapy, the median survival from the onset of GCM symptoms to death or transplant is only three months [158]. Immunosuppression therapy with steroids alone appears to provide no survival advantage although steroids in combination with additional immunosuppression therapy may improve survival but the optimal regimen is unknown [152]. The short-term use of muromonab-CD3 (a monoclonal antibody that binds to the T-cell receptor-CD3 complex on circulating T cells) followed by long-term cyclosporine and steroids leads to high one-year survival [156]. Clinical use of corticosteroid in addition to rabbit antithymocyte globulin also has had success in a small case series of GCM patients [162]. The use of multidrug immunosuppression, consisting predominantly of azathioprine, cyclosporine, and prednisone without T-cell depletion improves survival compared with historical controls with a five-year transplant-free survival of 52%. This survival is however inferior to long-term survival of patients without heart transplantation, with more than half of the surviving patients experiencing ongoing ventricular arrhythmias [153]. GCM patients are also more likely to require mechanical circulatory support often bi-ventricular as a bridge to transplantation compared with DCM patients [163,164]. In GCM patients who have undergone heart transplant, GCM may recur in 26% of patients after transplant [152].
Hypersensitivity myocarditis: Hypersensitive MC is a form of eosinophilic MC that is typically drug or vaccine related. It is rare, accounting for less than one percent of total unexplained cardiomyopathies [165]. The disease has a higher incidence in explanted heart of patients undergoing heart transplant but the high incidence may be explained by the use of dobutamine to bridge patients to transplant supported by multiple reports demonstrating dobutamine can cause drug-related eosinophilic CM [166,167]. Numerous drugs can be aetiologic agents of hypersensitivity MC and the disorder is often transient related to the inciting drug. In rare cases, MC can develop several years since the initiation of the inciting drug [168]. Clinical manifestations may be non-specific and the diagnosis is seldom suspected or established during life. Cardiac biopsy has a reasonably high yield in patients suspected of hypersensitive MC with eosinophilia although it may be also due to sampling error [165]. The ACCF/AHA and ESC scientific statement on the role of EMB in the management of CVD recommends EMB for patients with suspected allergic reaction or eosinophilia [169]. Peripheral eosinophilia accompanied by cardiac signs should raise suspicion, which is usually absent in the early stages of the disease. Treatment should target identifying and stopping the inciting agent and if possible, corticosteroids may be beneficial [10]. Smallpox vaccine may also cause myopericarditis with an eosinophilic and lymphocytic infiltrate and more than 10% of the patients will exhibit cardiac symptoms if chest pain, palpitations and dyspnoea within 30 days of vaccination, 0.4% will be diagnosed with clinical MC or pericarditis and 2-3% will have subclinical MC defined by elevated cardiac troponin serum levels [170,171].
Kawasaki disease: Kawasaki disease is the leading cause of acquired heart disease in children in developed countries [172]. Typically, Kawasaki disease affects infants and children under the age of five years as an acute febrile self-limiting mucocutaneous lymph node syndrome with multisystemic vasculitis [173-175]. Initially described in Japan in 1967 and reported in English in 1974, since then more than 116,000 cases have been reported in Japan although the disease is now known to have a worldwide distribution [175]. A subset of patients will go on to develop permanent damage to the arterial wall, valve leaflets and myocardium. The acute phase of the disease is self-limiting and diagnosis may be missed. If untreated, Kawasaki disease can lead to coronary aneurysm in 25% of the patients [176]. Patients experiencing coronary injury may develop thrombosis or stenotic lesions associated with aneurysms and are at a higher risk of myocardial infarction, SCD and HF [177,178].
Pathologic changes in the myocardium due to injury during the acute inflammatory phase have been previously reported as well as late complications such as residual injury from the acute vasculitis and MC [179]. In a study of the natural history of Kawasaki disease involving 594 Japanese subjects 10-12 years after disease onset, the reported long-term complications included stenosis, myocardial infarction, and death. Other case series report both symptomatic and asymptomatic coronary artery occlusion in regions of previous aneurysms [180,181], coronary artery stenosis [182], coronary artery calcification [183], diastolic dysfunction [184], and sudden death [185]. Diffuse MC followed by myocardial fibrosis may result in systolic and diastolic dysfunction in a subset of patients [186-189]. In the acute phase, tissue Doppler may reveal abnormal relaxation patterns associated with elevated levels of BNPs [187]. Decreased ventricular contractility and abnormal relaxation may result from antecedent myocardial inflammation and late-onset ventricular arrhythmias and HF [179,188].
Pathophysiology: Autoimmune CM has long been associated with and researched as an underlying aetiology of idiopathic DCM (iDCM). Ideally, AD is an umbrella term encompassing almost endless aetiologies and clinical presentations. Despite this diversity, ADs share many common clinical and pathological characteristics, which may include association with systemic and organ-specific inflammation, infections by microorganisms, genetic predisposition and correlation with specific human leukocyte antigen (HLA) subtypes [190].
Genetic predisposition: Genetic polymorphisms may predispose an individual to the development of ADs. The location of these polymorphisms are genes of proteins that regulate immune response such as major histocompatibility, complex proteins, immunoglobulins and T-cell receptor genes, and proteins involved in the metabolism of drugs, chemicals and antigens as well as within structural proteins themselves [190,191]. The exact genetic changes predisposing an individual towards an AD remain unidentified but comparison between concordance rates of AD in monozygotic twins and the rates of these ADs in the general population indicates that genetic factors may play a significant role in the development of ADs [192,193]. In addition to some of genetic predisposition involving unidentified mutations of sarcolemmal structural proteins such as lamin A/C, there appears to be a genetically derived propensity for ADs. Patients with iDCM are more likely to have another AD when compared to the general population and have a greater incidence of ADs such as psoriasis, thyroid disease, rheumatic arthritis and haemolytic anaemia in close family members of patients with iDCM [194].
HLA specificity: Human leukocyte antigen (HLA) genes, the human form of major histocompatibility complex (MHC) are among the most polymorphic genes in humans as well as the corresponding <MHC genes are in most mammals [9]. Evidenced based on familial prevalence and HLA distribution study, and validation study and a meta-analysis of published studies on HLA association links iDCM, and the HLA-DR4 and HLA-DQ4/6 subtypes. These HLA subtypes have a higher prevalence of 27% to 50% in patients with iDCM compared to 6% to 21% in non-DCM controls [195,196]. Both the HLA-DR and HLA-DQ subtypes are MHC class II proteins. After processing, these MHC class II proteins interact exclusively with CD4+ T-helper cells, whose primary mechanism of immunologic retaliation is the cytokine dependent activation of B-lymphocytes. Numerous cardiac-specific AABs have also been associated with iDCM [190].
Cardiac-specific AABs: Depending on the screening techniques used and type, the prevalence of cardiac specific AABs (mainly alpha myosin, beta1-adrenoreceptor, and cardiac troponin AABs) in patients with idiopathic is as high as 65% to 70%, many of which demonstrate significant pathologic potential [197-201].
α-myosin: AABs specific for α-myosin are formed through molecular mimicry of an infectious pathogen (mostly bacterial and virus) as well as are found in mice models of MC created by immunizing mice with α-myosin to mimic necrotic release of this protein. These α -myosin AABs contain epitopes responsible for producing the inflammatory response in murine model of MC [202] and occur in 23% to 66% of iDCM compared to 0.0% to 2.5% in healthy controls and their presence correlates with worsening LV systolic function and increased diastolic stiffness [197]. The persistence of α-myosin AABs correlates with increased serum levels of cardiac troponin to suggest persistent myocardial injury [203]. Recent evidence demonstrates α-myosin AABs cross-react with cell surface β-adrenoceptor to induce cyclic adenosine monophosphate-dependent phosphokinase-A activity [204]. Antibody-mediated activation of β-adrenoceptor induces cardiomyocyte apoptosis [205].
β1-Adrenoreceptor: β1-adrenoreceptor AABs are also formed through molecular mimicry of an infectious pathogen – specifically, AABs to a ribosomal protein of T. cruzi (Chagas disease) cross reacting with the second extracellular domain of the ß1-adrenoreceptor [206]. However, a majority of individuals with β1-adrenoreceptor AABs have not been infected with T. cruzi to suggest other mechanisms may underlie the production of β1-adrenoreceptor AABs. β1-adrenoreceptor AABs induces a dose-dependent increase in the rate of cardiomyocyte apoptosis, attenuated by the PKA inhibitor and the β selective antagonist metoprolol [205]. β1-adrenoreceptor occur in approximately 26% to 46% of patients with iDCM compared to 1% to 10% of healthy controls, and their presence correlates with cardiac specific morbidity and mortality [198,207-209]. Over a 10-year period, the presence of β1-adrenoreceptor has been shown to be an independent predictor of increased risk of all-cause and cardiovascular mortality [198] as well as is associated with depressed myocardial function and a higher incidence of SCD [201,204].
Cardiac troponin I: Nearly 50% of patients with severe iDCM exhibit increased serum levels of cardiac troponin [210]. Increased release of troponin may induce the formation of cardiac-specific AABs. Immunization of mice with cardiac troponin-I induced the formation of AABs, and the introduction of these AABs into healthy animals resulted in CM [211]. Although the same findings could not be produced by cardiac troponin, it could be explained by the location of cardiac troponin within cardiomyocytes whereas cardiac troponin-I on the outer cell membrane enabling it to interact with circulating AABs or lymphocytes [211,212]. A Japanese study evaluating the mechanism of cardiac troponin-I AABs induced myocardial damage reported that cardiac troponin-I AABs chronically increased L-type Ca2+ current via PKA-independent mechanism [212]. However, isolated cardiac troponin-I AABs from humans applied to culture rats myocytes did not induced any effect on Ca2+ currents nor binding to the cell surface [213].
Other cardia-specific AABs: Other cardiac-specific AABs include AABs specific for the sarcolemmal Na-K-ATPase, which is an independent predictor of poor systolic function, VT and SCD [214]. AABs specific M2-muscarinic acetylcholine receptor, which has been associated with a greater incidence of atrial fibrillation [199]. However, these findings may be inconclusive because of the small numbers of patients with these AABs and reported events in these studies, which warrants further work to fully evaluate their pathological potential.
Cellular autoimmunity: While research on humoral autoimmunity in iDCM patients has a disproportionate focus, there is only a paucity of information on cellular autoimmunity response. A CD4+ T lymphocyte response is an essential component in the production of antibodies. If the disease is infectious (viral), CD4+ is needed for the clearance of viral pathogens [190]. The involvement of CD4+ and CD8+ in the pathogenesis of iDCM lacks definitive evidence. However, indirect evidence such as histologic evaluation of endomyocardial biopsies of iDCM patients consistently reveal both CD4+ and CD8+ T-cell infiltrates [215,216]. In addition, injecting mice with lymphocytes from iDCM patients leads to the mice developing myocardial fibrosis and increased LV end-diastolic dimension, consistent with pathological changes in DCM [217].
Pathophysiologic mechanisms: The initiation of ADs occurs in the setting of a broken-down immune self-tolerance within a genetically susceptible environment [1]. In patients with iDCM, this breakdown in self-tolerance results from disease-associated necrotic release of immunologically sequestered cardiomyocyte proteins or by viral infection [218]. The breakdown typically manifests via four major pathways: (i) modification or release of immunologically sequestered proteins; (ii) polyclonal activation of lymphocytes; (iii) activation of resting T-cells via release of cytokines; or (iv) molecular mimicry [219]. The breakdown in self-tolerance may produce T-lymphocyte-mediated cellular autoimmune response and B-lymphocyte mediated humoral autoimmune response [218,219]. In T-lymphocyte mediated autoimmune response, cytotoxic CD8+ T-lymphocyte triggers the production of cytotoxic agents and subsequently induces apoptosis [220]. The activated cytotoxic CD8+ T-cells produce proliferative cytokines such as interlukin-2, resulting in expansion of the autoreactive clone and propagation of the autoimmune response. On the other hand, B-lymphocyte mediated autoimmune response, cellular antigens are processes by antigen presenting cells (macrophages, dendritic cells) and presented to CD4+ T cells, which subsequently active B-lymphocytes and induce the production of AABs specific to the self-antigen [221]. These AABs may be an inconsequential side-product of a cell-mediated autoimmune response or they may have directed deleterious effects on the cell of origin as in iDCM, in which the AABs response would also be responsible for propagation of the autoimmune response [219].
Meta-analysis of diagnosis and treatment: Autoimmune CM lacks specific diagnostic criteria and treatment strategies mainly because the disease has numerous aetiologies and a wide spectrum of clinical presentations that are non-specific [190]. Nevertheless, autoimmune CM shares several key clinicopathologic features that have a direct relevance to diagnosis and clinical management. The Systemic Lupus International Collaborating Clinics recommends the diagnosis of autoimmune CM should be by exclusion especially ruling out ischemic aetiology. Key diagnosis tests include (i) coronary angiography to exclude atherosclerosis, and (ii) echocardiography to assess cardiac function and structure in patients with signs and symptoms of HF [222]. The AHA scientific statement on treatment of different types of DCM recommends the first-line treatment for autoimmune conditions in patients with signs and symptoms of HF is immune modulating therapy that has no cardiotoxic effects. For patients with systolic HF, AHA recommends guideline-directed medical and device therapies and targeted treatment for patients with co-morbidities such as hypertension [23]. However, the current basis of diagnostic and treatment strategies for autoimmune CM is evidence-based strategies developed for iDCM and/or MC. Evidence specific for autoimmune CM is sporadic and largely derived from case reports and series, and non-randomized prospective and retrospective cohort studies. This meta-analysis therefore seeks to pool together published evidence on diagnosis and treatment of autoimmune CM to determine the most common clinical features, their evaluation and treatment strategies.
A PubMed search using a combination of the following keywords: “autoimmune disease”, “autoimmune cardiomyopathy”, “inflammatory cardiomyopathy”, “diagnosis” and “treatment” yielded two hundred and thirty-four (234) articles. Of these, 22 articles were included as highly relevant to this systematic review and meta-analysis. Articles published between 2000 and 2019 were included in this review, the period consistent with an era of increased research effort in the role of autoimmunity in the pathogenesis of CM. The included articles were limited to cases of diagnosis and clinical management of autoimmune CM. Excluded articles were case series, conference papers, review articles and studies with insufficient information to satisfy the aim of this review. Categorical data was presented as frequency and percentage, continuous variables as event rate and 95% confidence intervals (CI), and dichotomous data as weighted mean difference and 95% CI. The I2 statistics was used to quantify the percentage of total variation across studies that is not due to chance. Fixed effect model was used when I2 ≥ 50% while random effect model was used when I2 < 50% and a p-value of < 0.05 was considered statistically significant (Table 1).
Table 1. Summary of included studies (E/A: Ratio between peak early (E), and late (A) myocardial diastolic velocities; ECG: Electrocardiogram; Echo: Echocardiograph; LAD: Left Atrial Diameter; LV: Left Ventricular; LVEDD: Left Ventricular End-Diastolic Diameter; LVESD: Left Ventricular End-Systolic Diameter; PWT: Posterior Wall Thickness; RA: Rheumatoid Arthritis; SLE: Systemic Lupus Erythematosus; STE: Speckle Tracking Echocardiography; SWT: Septal Wall Thickness; TDI: Tissue Doppler Imaging; TTE: Transthoracic Echocardiography)
Author [Ref #] |
Year |
Design |
No. |
Women (n) |
Age (SD) |
Cause |
Test |
A summary of the main findings |
Tlustochowicz [223] |
1995 |
Prospective |
70 |
53 |
56.7 (11.2) |
RA |
ECG |
Cardiac arrhythmias occur in 50% of RAs patients and is independent of disease progression and type of treatment. |
Chapelon-Abric [224] |
2004 |
Retrospective |
41 |
18 |
38 |
CS |
ECG, echo |
ECG is abnormal in 68% consisting of conduction disturbances (43%) and arrhythmias (46%), and echo is abnormal in 77%. |
Forsblad-d’Elia [225] |
2013 |
Cross-sectional |
210 |
90 |
49 (13) |
AS |
ECG |
Cardiac conduction abnormalities occur in 10-33% of patients with AS depending on the diagnostic criteria. |
Touma [226] |
2013 |
Prospective |
274 |
243 |
47.7 (14.0) |
SLE |
ECG |
ECG abnormalities occur in 40.5%: ST-T changes in 19.7%, LVH in 9.9%, BBB in 4.7% and pathological Q waves in 6.2%. |
Bacani [227] |
2015 |
Prospective |
813 |
556 |
55.9 (15.7) |
RA |
ECG |
AF occurs in 4% of RA patients with a higher cumulative incidence (18.3%) after adjusting for age, sex, smoking, and hypertension. |
Bourre‐Tessier [228] |
2015 |
Prospective |
779 |
687 |
35.2 (13.8) |
SLE |
ECG |
Common ECG abnormalities – ST-T changes (30.9%), LVH (5.4%), arrhythmias (1.3%), and QTc ≥140 msec (15.3%) |
Chauhan [229] |
2015 |
Prospective |
518 |
354 |
58.5 (15.1) |
RA |
ECG |
Elevated risk of QTC prolongation – at index date 15% and cumulative incidence after 20 years 46%, with marginal association with all-cause mortality. |
Geraldino-Pardilla [230] |
2016 |
Cross-sectional |
50 |
46 |
36.0 (13.0) |
SLE |
ECG |
Longer QTc (by 26ms) and 3.3 fold higher prevalence of non-specific ST-T changes (56% vs. 17%) in SLE than RA patients |
|
|
|
139 |
85 |
59.0 (8.0) |
RA |
ECG |
Masooleh [231] |
2016 |
Prospective |
100 |
77 |
49.74 (11.56) |
RA |
ECG, echo |
ECG abnormalities occurred in 32%, ST-T changes was the most common (15%). Echo abnormalities occurred in 74%, pericardial (47%) and ventricular dysfunction (45%) being the most common. |
Al Rayes [232] |
2017 |
Prospective |
487 |
390 |
30.5 (11.9) |
SLE |
ECG |
High prevalence (24.2%) of ECG abnormalities supporting the need for early identification of ECG changes |
El-Monem [233] |
2019 |
Prospective |
30 |
27 |
37.9 (6.5) |
RA |
ECG |
ECG changes were present in 33.3% of RA patients consisting of ST-T changes (23%), and LVH (10%), and in 90% of patients using steroids. |
|
|
|
30 |
28 |
34.7 (7.9) |
SLE |
ECG |
ECG changes were present in 10% of SLE patients, consisting of ST-T changes (66.7%) and LBBB (33.3%) |
Di Franco [234] |
2000 |
Prospective |
32 |
28 |
50 (10) |
RA |
Echo |
RA patients have LV filling abnormalities characterized by reduced E/A ratio (1.16±0.31) compared to controls (1.37±0.32) and raised pulmonary venous flow ratio (1.43±0.4 vs. 1.22±0.29) |
Guedes [235] |
2001 |
Prospective |
30 |
26 |
57.8 (15.1) |
RA |
ECG, Echo |
Cardiac involvement of the MR is extremely common (80%) in RA patients compared to controls (37%). |
Hameed [236] |
2007 |
Prospective |
48 |
44 |
26 |
SLE |
Echo |
Echo abnormalities were present in 58.3% of RA patients involving pericardial (57%), valvular (43%) and myocardial (43%) |
Allam [237] |
2013 |
Prospective |
50 |
50 |
26.82 (6.96) |
SLE |
Echo |
Tissue Doppler velocities are a sensitive tool in detecting early myocardial dysfunction when 2D echo findings are normal |
Bakhoum [238] |
2015 |
Cross-sectional |
50 |
47 |
28.5 (8.5) |
SLE |
Echo |
Medial and lateral mitral annulus velocities are significantly lower. Global longitudinal and circumferential were significantly lower |
Klingberg [239] |
2015 |
Prospective |
187 |
82 |
59.0 (11.3) |
RA |
Echo |
18% had aortic regurgitation and 13% had conduction system abnormalities associated with disease duration and patient’s age. |
Yazici [240] |
2015 |
Prospective |
200 |
45 |
57.12 (12.08) |
RA |
Echo |
6% had systolic dysfunction, 4,5 pericardial infusion, 8.5% valvular dysfunction, 68.1% diastolic dysfunction. |
Mathew [241] |
2017 |
Cross-sectional |
99 |
90 |
36.6 (10.7) |
RA=55; SLE=32; CTD=12 |
ECG, echo, chest x-ray |
ECG abnormalities were present in 27% of patients (sinus bradycardia/tachycardia, ST-T changes) and echo abnormalities in 32 % and cardiomegaly in 10% |
Hannawi [242] |
2018 |
Cross-sectional |
39 |
34 |
NR |
RA |
Echo |
RA patients have a higher LVED and LEVSD and greater LV mass index compared to healthy controls (p < 0.05). |
Mohammed [243] |
2018 |
Prospective |
50 |
44 |
27.7 (8.5) |
SLE |
Echo |
High frequency of mitral (64%), aortic (22%) and tricuspid (24%) regurgitation, and pericardial effusion (22%), and LVH (10%) |
Mohamed [244] |
2019 |
Cross-sectional |
59 |
51 |
31.3 (10.5) |
SLE |
Echo |
Mitral (33.9%) and tricuspid (32.2%) regurgitation, and pericardial effusion (13.6%), |
Findings
Twenty-two (22) studies published between 1995 and 2019 investigating autoimmune diseases with cardiac involvement were included in the present review [223-244]. The studies consisted of fifteen (15) prospective studies [223,226-229,231-237,239,240,243], six (6) cross-sectional studies [225,230,238,241,242,244] and one (1) retrospective study [224] (Table 1). In total, the 22 studies enrolled 4,385 patients comprising of a greater majority of women (n = 3,195; 73%) and mean age 43 years range 26-59. Except one study [224] that enrolled 41 cardiac sarcoidosis patients (0.09%) (Granulomatous disease), all the remaining 21 enrolled 4,344 patients (99.01%) with CTDs (SLE, RA and AS). Two studies enrolled SLE and RA patients [230,233] and each patient group was analysed separately. The mean duration of the disease was 10.18 years, range 4.1 to 24.3 years. The most common diagnostic test used in 13 studies each were ECG [223-233,235,241] and echocardiography [224,231, 234-244], and less commonly chest x-ray in used in two studies [235,241].
ECG is a routine test recommended for all patients suspected with CM. In a pooled analysis of eight studies [223,224,226,227,231,231,233,241] ECG abnormalities were present in 433 out of 1,944 patients translating into an event rate of 31.8% (95% CI: 17.6% to 50.4%) (Figure 1). The most common ECG abnormality was ST-T changes present in 516 of 2,450 patients with an event rate (ER) of 18.8% (95% CI: 13.7% to 25.4%; p = 0.000) pooled from nine (9) studies [224,226,228-234] (Figure 2). LVH was common in 156 out of 2,161 translating into an incidence of 7.5% (95% CI: 6.4% to 8.7%; p = 0.000) (Figure 3). Other ECG abnormalities included BBB present in 138/1,471 patients (ER: 5.7%; 95% CI: 3.2% to 9.9%) pooled from nine (9) studies [224,225,226,228,229,231,232,233,234] (Figure 4), and arrhythmias present in 175/2,635 patients (ER: 5.0%; 95% CI: 2.6% to 9.4%; p = 0.000 pooled from six (6) studies [224-229] (Figure 5).
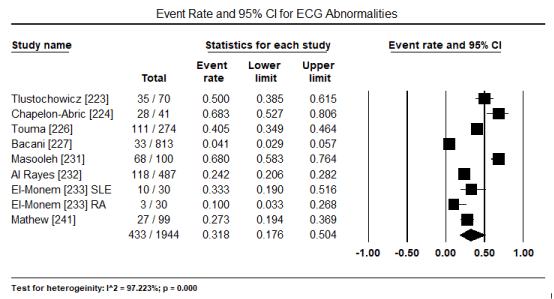
Figure 1. Forest plot for event rate for overall ECG abnormalities
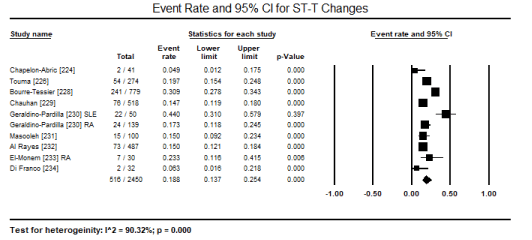
Figure 2. Forest plot for event rate for ST-T changes
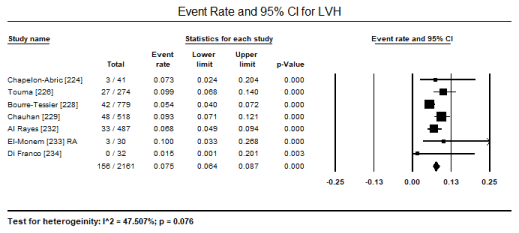
Figure 3. Forest plot for event rate for LVH
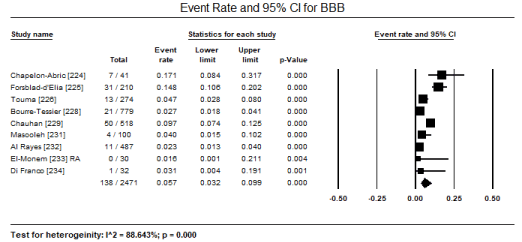
Figure 4. Forest plot for event rate for bundle branch block
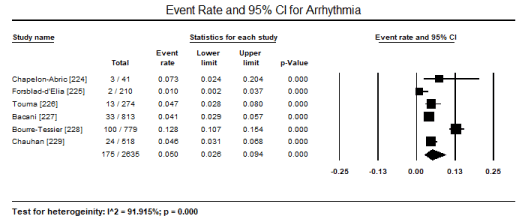
Figure 5. Forest plot for event rate for arrhythmia
Transthoracic echocardiography (TTE) and tissue Doppler imaging (TDI) were the most common non-invasive modality used for assessing abnormalities in cardiac structure and function. In five studies [224,235,236,241,244], echocardiography abnormalities were present in 152/277 patients translating into an event rate of 63.6% (95% CI: 43.0% to 80.1%) (Figure 6). The most frequently observed changes compared to control group were significantly reduced E/A ratio, (weighted mean difference [WMD] -0.156; 95% CI: -0.264 to -0.049; p = 0.004) (Figure 7); and significantly increased LVESD (WMD: 2.15; 95% CI: 0.52 to 4.61; p= 0.001) (Figure 8); and LVEDD (WMD 2.57; 95% CI: 0.52 to 4.61; p= 0.01) (Figure 9) pooled from three studies [234,237,238]. TDI also showed a slight but significant (p<0.05) decrease in LV septal wall thickness (SWT) and posterior wall thickness (PWT) [237,238]. When standard echocardiography is inconclusive, speckle tracking echocardiography (STE) can reveal significant subclinical changes in cardiac function in septum and lateral mitral annulus (p < 0.05) [237] and in global longitudinal strain (GLS) and global circumferential strain (GCS; p < 0.001) [238].
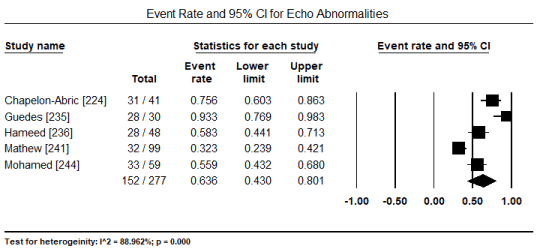
Figure 6. Forest plot for event rate for echo abnormalities
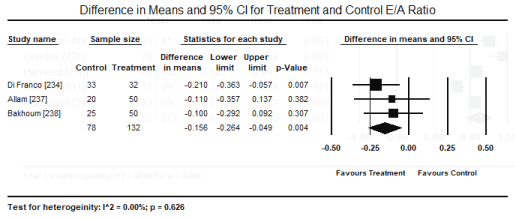
Figure 7. Forest plot for difference in means for E/A ratio
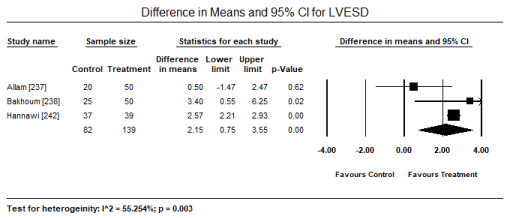
Figure 8. Forest plot for difference in means for LVESD
Figure 9. Forest plot for difference in means for LVEDD
Numerous diseases capable of causing autoimmune CM and marked differences in the type and frequency of myocardial involvement render the development of a general diagnostic algorithm and treatment strategy too cumbersome to be clinically useful. Consequently, it is a common practice for current consensus guidelines for autoimmune CMs to adopt a disease-specific approach. Despite numerous aetiology, autoimmune CMs shares several general characteristics and pathophysiologic mechanisms whose understanding would be clinically helpful to raise the clinical suspicion of the disease as well as refine clinical management. In the present meta-analysis, it was apparent that studies specific to autoimmune CM are lacking with a greater majority enrolling ADs patients with cardiac involvement that includes pericarditis and valvular diseases. Moreover, much evidence regarding autoimmune CMs come from studies on CTDs, with fewer studies focussing on granulomatous or inflammatory diseases. The studies included in the present meta-analysis provide an important insight into the general diagnostic and treatment features of patients suspected to have autoimmune CM.
Diagnosis
The present analysis lacked sufficient data for a pooled analysis of clinical signs and symptoms. Generally, early diagnosis of autoimmune CM is challenging because of non-specific clinical signs and symptoms. However, clinical signs and symptoms such as unexplained dyspnoea, palpitations, chest pain, syncope, arrhythmia, HF aborted SCD and cardiogenic shock may be helpful to raise clinical suspicion of the disease [241]. In many ADs, coronary artery disease (CAD) and coronary microvascular dysfunction may contribute to cardiac signs and symptoms and thus excluding ischemic aetiology is an important initial test. Elevated cardiac biomarkers such as troponin and/or NT-pro-BNP may indicate myocardial involvement irrespective of the aetiology. However, cardiac biomarkers are non-specific since troponin may be increased in extra cardiac disease such as pulmonary embolism and MC may occur in the absence of troponin. Nevertheless, disease specific biomarkers such as AABs are available for various ADs and form part of multi-parametric diagnostic criteria, but up to now, it remains unknown whether cardiac biomarkers have an association with MC [245]. Clinical signs and symptoms, and cardiac biomarkers form part of patient selection in the included studies but the data was insufficient to support a pooled analysis and thus the focus was on ECG and echocardiographic evaluation.
Generally, ECG is a routine test recommended for initial diagnosis of heart disease and echocardiographic is a common non-invasive imaging test for abnormalities in cardiac structure and function. The present findings reveal a high prevalence of ECG and echo abnormalities in 31% and 64% respectively of patients with autoimmune CMs. Common ECG abnormalities include ST-T changes (18.8%), LVH (7.5%), BBB (5.7%) and arrhythmias (5.0%). Echocardiographic modalities – TTE and TDI – are the most commonly used for assessing cardiac structural and functional parameters. Compared to healthy controls, patients with autoimmune CMs exhibit significantly reduced E/A ratio and increased LVESD and LVED. STE is also an important parameter to assess for early myocardial changes using GLS and GCS parameters before the appearance of noticeable changes in ventricular structure on standard echocardiography. These findings reinforce previous studies as well as recommendations published in a position statement of the ESC working group on myocardial and pericardial diseases on the diagnosis and management of myocardial involvement in patients with ADs [245]. Although ECG abnormalities are non-specific, the ESC underscores the importance of routine ECG test in the diagnosis of autoimmune CM because ECG is a useful test to raise clinical suspicion of cardiac involvement to warrant additional non-invasive imaging tests to confirm autoimmune CM.
Although the ESC recommends the role of non-invasive imaging in detecting myocardial involvement, the findings are usually non-specific to the underlying aetiology [245]. The first-line modality for non-invasive imaging is standard echo (TTE or TEE) with Doppler analysis for the diagnosis of MC, pericarditis and valvular involvement [246-248]. Advanced echocardiographic methods such as STE may be used to detect subclinical myocardial development when the findings of TTE or TEE are inconclusive. TTE is useful in particular cases such as Libman-Sacks endocarditis in SLE [249,250]. Evaluation of tricuspid and pulmonary regurgitation gradients is a useful finding for non-invasive diagnosis of pulmonary hypertension [250,251]. In cases where TTE and TEE tests are negative or non-specific, STE findings of significantly reduced GLS and GCS are subclinical changes suggesting the early onset of autoimmune CM [237,238].
Although the present analysis did not assess the diagnostic value of cardiac MRI in patients with autoimmune CM, consensus reports suggest that cardiac MRI with tissue characterization sequences and positron emission tomography (PET) may refine clinical suspicion of non-ischemic inflammatory myocardial involvement and helpful in patient follow-up and assessing response to treatment. Cardiac MRI may also be helpful to provide complementary information and is particularly useful when results of echocardiography are inconclusive [245]. Cardiac MRI tissue characterization with T1- and T2-weighted imaging and late gadolinium enhancement (LGE) as well as parametric mapping can be useful in assessment of cardiac involvement in ADs. Characteristic sub-epicardial or mid-myocardial cardiac MRI LGE pattern in ADs allows differential diagnosis from coronary artery disease and its findings correlates with disease activity in patients with RA and systemic sclerosis [252,253].
Finally, endomyocardial biopsy (EMB) is the gold standard for evaluating patients suspected with MC with or without ADs although the current use of EMB is limited to specialized centers. Using histological, immunological, immunohistochemical and molecular tools, EMB can differentiate between infectious and non-infectious MC. EMB is useful for the identification of cardiac vasculitis and other non-inflammatory (degenerative or infiltrative) myocardial diseases [169,245,255-257]. EMB is particularly useful for the diagnosis of ADs if cardiac clinical and non-invasive findings suggest non-ischemic myocardial involvement as well as in the presence of unexplained and unexpected change in cardiac status and the histological confirmation is required to guide a change in its management. This is especially true if the diagnosis cannot be made with biopsies of more accessible tissues such as in cardiac amyloidosis. The drawback of EMB is high complications rate for inexperienced clinicians and sampling errors. In the case of histology and immunochemistry, the tests require at least three myocardial tissues sampled from the RV or LV [245,254].
Therapeutics
Clinical management of autoimmune CM is one of the less developed areas in the MC and CM field. To begin with, some clinical studies lack a clear diagnostic and aetiologic definition generating a conundrum on how to analyse the results of therapeutic trials. It also remains unknown at which point of the temporal evolution of the ADs process are the patients when enrolled in therapeutic trials to support the hypothesis that case and control groups are heterogeneous in immunopathogenic terms. The potential severity and relatively low incidence of autoimmune CM along with immunologic and diagnostic complexities or limitations enounced above make it almost impossible to conduct optimally designed clinical trials as the ones performed for other high incidence entities such as high blood pressure and diabetes [19]. Because of different types and frequencies of myocardial involvement in different ADs, it was not possible for the present meta-analysis to pool analysis. There is also a lack of robust evidence-base for clinical management of affected patients and myocardial inflammatory involvement is often under-diagnosed and overlooked. The ESC statement on the management of MC recommends disease specific therapies in ADs patients that includes immunosuppressive and/or immunomodulatory regimes, particularly in the active phase of the disease [259,260]. Presently, a growing body of therapeutic knowledge strongly suggests that immune modulation or suppression has beneficial effects in the clinical management of MC [261,262].
Immunosuppression
The rationale behind immunosuppression therapy is targeting pro-inflammatory mediators of the disease. Immunosuppression has proved effective in the treatment of autoimmune MC such as giant cell and hypersensitive MC. The use of broad immunosuppressive drugs instead of targeted treatment have been used. Some of the key drugs tested for autoimmune MC include steroids (prednisone, prednisolone), cyclosporine and azathioprine [152,263]. Although, these drugs are often used in combination, there is a need for proper clinical comparative studies to evaluate the benefits for individual subtypes of autoimmune CM. Current evidence on the efficacy of immunosuppression therapy is inconclusive. While the TIMIC study reported significant or at least some improvement in terms of ejection fraction [264], the Myocarditis Treatment Trial found no benefit on ejection fraction or survival compared to placebo [265]. However, the inconsistent findings can be attributable to differences in patient selection. The study reporting benefit enrolled non-viral MC whereas the study reporting no benefit did not make any discrimination on patients. Another prospective study of 12 GCM patients analysing the effect of monoclonal antibody targeting CD3 (muromonab-CD3) in combination with cyclosporine and steroids showed improved survival [266]. Treatment complications of immunosuppression include increased likelihood of acquired infection and/or reactivation of latent or opportunistic infection. In particular, the use of corticosteroids has been associated with adverse CVD outcomes and unfavourable metabolic profiles [267]. Thus, steroid-sparing strategies are preferred especially if chronic immunosuppression is needed [260]. Due to some teratogenic and oncogenic effects of some immunosuppressants such as methotrexate or mycophenolate mofetil, there might be a need to minimize usage and active screening programs advisable in high-risk populations [245].
Immunomodulation
Consistent findings from several studies associate immunoadsorption co-administered with immunoglobulin with improved hemodynamic particularly improved ejection fraction [268-271]. These studies enrolled DCM patients, which support the hemodynamic deleterious effect of cardiac AABs as well as DCM patients of any cause, which expands the usefulness of immunomodulation to non-MC patients. High doses of immunoglobulin exerts a beneficial effect – improvement in survival and ejection fraction in patients with acute MC [262,271]. However, one controlled study did not find any improvement in ejection fraction although it did not make a clear aetiologic definition [272]. Initially, intravenous immunoglobulin (IVIG) was an effective treatment of a series of disease of known immunologic origins such as Kawasaki syndrome, dermatomyositis, multiple sclerosis, idiopathic thrombocytopenic purpura and Guillain-Barre syndrome [18]. Recently, its efficacy has been proven in the treatment of recent onset of MC and myocardiopathy in children. In a retrospective study of 21 children diagnosed with MC (with or without EMB evidence of inflammatory infiltrate) and treated with IVIG, there was a marked improvement in LVEF during the first weeks or treatment and maintained at 1-year follow-up compared to controls who did not receive IVIG [273].
Immunoadsorption (apheresis)
Apheresis refers to the use of various therapeutic extracorporeal purification techniques to eliminate high-molecular weight substances in blood. Plasmapheresis and immunoadsorption are the most frequently used techniques. Plasmapheresis is a non-selective extracorporeal technique for eliminating toxic elements in blood while immunoadsorption is a selective apheresis technique that attempts to eliminate human immunoglobulin extracorporeal by using specific immunoadsorbent substances [18]. Both techniques involve the extraction of blood, separation of the plasma from form elements and reinfusion of the patient with these substances while the difference is plasmapheresis is non-selective while immunoadsorption selectively eliminates specific substances. The hypothesis behind apheresis is that the presence of AABs against specific beta-receptor epitopes in patients with iDCM, and that, AABs could play a determining role in the pathophysiology of illness. Thus, the elimination of the causative agent might result in substantial improvement in illness. The decrease in AAB titre after the administration of immunosuppressive therapy or the extraction of ABBs from serum might improve both contractile myocardial function and the clinical course of illness [18]. Muller et al. [201] performed a retrospective study with a control group and reported significant improvement in LV function and NYHA functional class after 1-year follow-up. However, additional trials with a larger sample with more extensive follow-up periods are warranted to confirm beneficial effects of apheresis therapy.
Clinical implications
Due to differences in the type and frequency of myocardial involvement in autoimmune CM, future directions should focus on proper diagnostics on aetiology as well as on the specific stage of autoimmune process. Such a diagnosis strategy should allow the determination of specific treatment depending on the underlying cause and on the predominant immune process occurring in the patient at the time of intervention. In the near future, there may be a need to develop targeted treatment strategy such as managing the specific immunopathogenic processes predominating in the patient including inducing self-tolerance, boosting of Treg response and reducing Teff response, blocking innate response-associated receptors, and modulation of immunologic role of the stromal cells such as fibroblasts and endothelial cells. Expanding knowledge about immunopathogenesis of MC may end up in the development of effective diagnostic, prognostic and therapeutic strategies [245]. In future, it may also be practical to separate autoimmune CM from iDCM to improve diagnosis and clinical management.
Autoimmune diseases (ADs) are conditions triggered by the immune system initiating an attack on self-molecules due to the deterioration of immunologic tolerance to autoreactive immune cells. Genetics, infections and environmental factors may play a role in the initiation of ADs. Connective tissue, granulomatous and inflammatory diseases are common causes of non-infectious autoimmune CM. Key clinicopathological characteristics are genetic predisposition, HLA specificity, cardiac-specific AABs and cellular autoimmunity. Pathophysiologically, the initiation of autoimmune CM occurs after the breakdown of immune self-tolerance within a genetically susceptible setting. Upon the establishment of the initial myocardial inflammation, T-cell dependent autoimmune process occurs despite difference in the specific aetiologic factor. However, specific triggers and factors contributing to the progression to autoimmune CM remains a conundrum, since no risk or aetiologic factor seems to be sufficient for the initiation and progression from MC to CM. General diagnostic algorithm is lacking largely due to heterogeneous aetiology, and the current knowledge of cardiac manifestation stems largely from clinical and echocardiography studies, which are non-specific to confirm diagnosis. Despite advances in therapeutics, treatment still relies on global immunosuppression and unspecific immunomodulation therapy usually with suboptimal results. The current expectation is that future clinical trials should investigate and provide deeper insights into the pathogenesis of autoimmune CM, which would lead to the development of better diagnostic tools to allow the characterization and stratification of the stages of autoimmune CM as well as the development of individualized targeted treatments.
- Viswanath D (2013) Understanding autoimmune diseases: a review. Journal of Dental and Medical Sciences 6: 08-15.
- Singh AP, Wal P, Wal A, Srivastava V, Tiwari, R et al. (2016) Understanding autoimmune disease: an update review. International Journal of Pharmaceutical Technology and Biotechnology 3: 51-65.
- Wang L, Wang FS, Gershwin ME (2015) Human autoimmune diseases: a comprehensive update. J Intern Med 278: 369-395. [Crossref]
- Jacobson DL, Gange SJ, Rose NR, Graham NM (1997) Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol 84: 223-243. [Crossref]
- Eaton WW, Rose NR, Kalaydjian A, Pedersen MG, Mortensen PB (2007) Epidemiology of autoimmune diseases in Denmark. J Autoimmun 29: 1-9. [Crossref]
- Smith DA, Germolec DR (1999) Introduction to immunology and autoimmunity. Environ Health Perspect. 107: 661-665. [Crossref]
- Kindermann I, Barth C, Mahfoud F, Ukena C, Lenski M et al. (2012) Update on myocarditis. J Am Coll Cardiol 59: 779-792. [Crossref]
- Kindermann I, Kindermann M, Kandolf R, Klingel K, Bultmann B et al. (2008) Predictors of outcome in patients with suspected myocarditis. Circulation 118: 639-648. [Crossref]
- Bracamonte-Baran W, Čiháková D (2017) Cardiac autoimmunity: myocarditis. Adv Exp Med Bio 1003: 187-221. [Crossref]
- Trachtenberg BH, Hare JM (2017) inflammatory cardiomyopathic syndromes. Circ Res 121: 803-818. [Crossref]
- Mackay IR, Leskovsek NV, Rose NR (2008) Cell damage and autoimmunity: a critical appraisal. J Autoimmun 30: 5-11. [Crossref]
- Bracamonte-Baran W, Burlingham W (2015) Non-inherited maternal antigens, pregnancy, and allotolerance. Biom J 38: 39-51. [Crossref]
- Von Herrath MG, Oldostone MBA (1996) Virus-induced autoimmune disease. Curr Opin Immunol 8: 878-885. [Crossref]
- Mountz JD, Talal N (1993) Retrovirus, apoptosis and autogenes. Immunol Today 14: 532-536. [Crossref]
- Yoshida S, Gershwin E (1993) Autoimmunity and selected environmental factors of disease induction. Semin Arthritis Rheum 22: 399-419. [Crossref]
- Griem P, Gleichmann E (1995) Metal ion induced autoimmunity. Curr Opin Immunol 7: 831-838. [Crossref]
- Lanzavecchia A (1995) How can crytic epitopes trigger autoimmunity? J Exp Med 181: 1945-1948. [Crossref]
- San Martín MA, García Á, Rodríguez FJ, Terol I (2002) Dilated cardiomyopathy and autoimmunity: an overview of current knowledge and perspectives. Rev Esp Cardiol 55: 514-524. [Crossref]
- Fu M, Matsui S (2005) Is cardiomyopathy an autoimmune disease? Keio J Med 51: 208-212. [Crossref]
- Neumann DA (1994) Autoimmunity and idiopathic dilated cardiomyopathy. Mayo Clin Proc 69: 193-195. [Crossref]
- Yoshikawa T, Baba A, Nagatomo Y (2009) Autoimmune mechanisms underlying dilated cardiomyopathy. Circ J 73: 602-607[Crossref]
- Caforio AL, Bottaro S, Iliceto S (2012) Dilated cardiomyopathy (DCM) and myocarditis: classification, clinical and autoimmune features. Appl Cardiopulm Pathophysiol.16: 82-95.
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646. [Crossref]
- Chaigne B, Scirè CA, Talarico R, Alexander T, Amoura Z et al. (2019) Mixed connective tissue disease: state of the art on clinical practice guidelines. RMD Open 4: e000783. [Crossref]
- Di Battista M, Marcucci E, Elefante E, Tripoli A, Governato G et al. (2018) One year in review 2018: systemic lupus erythematosus. Clin Exp Rheumatol 36: 763-777. [Crossref]
- Murphy G, Lisnevskaia L, Isenberg D (2013) Systemic lupus erythematosus and other autoimmune rheumatic diseases: challenges to treatment. Lancet 382: 809-818. [Crossref]
- Bjornadal L, Yin L, Granath F, Klareskog L, Ekbom A (2004) Cardiovascular disease a hazard despite improved prognosis in patients with systemic lupus erythematosus: results from a Swedish population based study 1964-1995. J Rheumatol 31: 713-719. [Crossref]
- Schoenfeld SR, Kasturi S, Costenbader KH (2013) The epidemiology of atherosclerotic cardiovascular disease among patients with SLE: a systematic review. Semin Arthritis Rheum. 2013; 43:77-95. [Crossref]
- Zawadowski GM, Klarich KW, Moder KG, Edwards WD, Cooper LT Jr (2012). A contemporary case series of lupus myocarditis. Lupus 21:1 378-1384. [Crossref]
- Ward MM (1999) Premature morbidity from cardiovascular and cerebrovascular diseases in women with systemic lupus erythematosus. Arthritis Rheum 42: 338-346. [Crossref]
- Urowitz MB, Gladman D, Ibanez D, Bae SC, Sanchez-Guerrero J et al. (2010) Systemic Lupus International Collaborating Clinics. Atherosclerotic vascular events in a multinational inception cohort of systemic lupus erythematosus. Arthritis Care Res (Hoboken) 62: 881-887. [Crossref]
- Jain D, Halushka MK (2009) Cardiac pathology of systemic lupus erythematosus. J Clin Pathol 62: 584-592. [Crossref]
- O’Neill S, Cervera R (2010) Systemic lupus erythematosus. Best Pract Res Clin Rheumatol 24: 841-855. [Crossref]
- Wijetunga M, Rockson S (2002) Myocarditis in systemic lupus erythematosus. Am J Med 113: 419-423. [Crossref]
- Nord JE, Shah PK, Rinaldi RZ, Weisman MH (2004) Hydroxychloroquine cardiotoxicity in systemic lupus erythematosus: a report of 2 cases and review of the literature. Semin Arthritis Rheum 33: 336-351. [Crossref]
- Ratliff NB, Estes ML, Myles JL, Shirey EK, McMahon JT (1987) Diagnosis of chloroquine cardiomyopathy by endomyocardial biopsy. N Eng J Med 316: 191-193. [Crossref]
- Soong TR, Barouch LA, Champion HC, Wigley FM, Halushka MK (2007) New clinical and ultrastructural findings in hydroxychloroquine-induced cardiomyopathy: a report of 2 cases. Hum Pathol 38: 1858-1863. [Crossref]
- Paradiso M, Gabrielli F, Masala C, Coppotelli L, Di Franco M et al. (2001) Evaluation of myocardial involvement in systemic lupus erythematosus by signal-averaged electrocardiography and echocardiography. Acta Cardiol 56: 381-386. [Crossref]
- Buss SJ, Wolf D, Korosoglou G, Max R, Weiss CS et al. (2010) Myocardial left ventricular dysfunction in patients with systemic lupus erythematosus: new insights from tissue Doppler and strain J Rheumatol 37: 79-86. [Crossref]
- Mavrogeni S, Bratis K, Markussis V, Spargias C, Papadopoulou E et al. (2013) The diagnostic role of cardiac magnetic resonance imaging in detecting myocardial inflammation in systemic lupus erythematosus: differentiation from viral myocarditis. Lupus 22: 34-43. [Crossref]
- Doherty NE, Siegel RJ (1985) Cardiovascular manifestations of systemic lupus erythematosus. Am Heart J 110: 1257-1265. [Crossref]
- Naarendorp M, Kerr LD, Khan AS, Ornstein MH (1999) Dramatic improvement of left ventricular function after cytotoxic therapy in lupus patients with acute cardiomyopathy: report of 6 cases. J Rheumatol 26: 2257-2260. [Crossref]
- Sherer Y, Levy Y, Shoenfeld Y (1999) Marked improvement of severe cardiac dysfunction after one course of intravenous immunoglobulin in a patient with systemic lupus erythematosus. Clin Rheumatol 18: 238-240. [Crossref]
- Disla E, Rhim HR, Reddy A, Ramaprasad S, Taranta A (1993) Reversible cardiogenic shock in a patient with lupus myocarditis. J Rheumatol 20: 2174. [Crossref]
- Ibanez B, James S, Agewall S, Antunes MJ, Bucciarelli-Ducci C et al. (2018) 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J 39: 119-177. [Crossref]
- Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG et al. (2016) 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Eur Heart J 37: 2129-2200. [Crossref]
- McInnes IB, Schett G (2011) The pathogenesis of rheumatoid arthritis. New Eng J Med 365: 2205-2219. [Crossref]
- Voskuyl AE (2006) The heart and cardiovascular manifestations in rheumatoid arthritis. Rheumatology (Oxford) 45: iv4-iv7. [Crossref]
- Goldenberg J, Ferraz MB, Pessoa AP, Fonseca AS, Carvalho AC et al. (1992) Symptomatic cardiac involvement in juvenile rheumatoid arthritis. Int J Cardiol 34: 57-62. [Crossref]
- Nicola PJ, Maradit-Kremers H, Roger VL, Jacobsen SJ, Crowson CS et al. (2005) The risk of congestive heart failure in rheumatoid arthritis: a population-based study over 46 years. Arthritis Rheum 52: 412-420. [Crossref]
- Wright K, Crowson CS, Gabriel SE (2014) Cardiovascular comorbidity in rheumatic diseases: a focus on heart failure. Heart Fail Clin 10: 339-352. [Crossref]
- Hurd ER (1979) Extraarticular manifestations of rheumatoid arthritis. Semin Arthritis Rheum. 8: 151-176. [Crossref]
- Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE et al. (2013) 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Col Cardiol 62: e147-239. [Crossref]
- Guillevin L, Lhote F (1995) Polyarteritis nodosa and microscopic polyangiitis. Clin Exp Immunol 101: 22. [Crossref]
- Guillevin L. Polyarteritis Nodosa (2017) In the heart in rheumatic, autoimmune and inflammatory. Diseases 419-427. Academic Press.
- Pagnoux C, Seror R, Henegar C, Mahr A, Cohen P et al. (2010) French Vasculitis Study Group. Clinical features and outcomes in 348 patients with polyarteritis nodosa: a systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French Vasculitis Study Group Database. Arthritis Rheum 62: 616-626. [Crossref]
- Ramalho AR, Costa S, Silva F, Donato P, Franco F et al. (2017) Autoimmune myocarditis in systemic sclerosis: an unusual form of scleroderma heart disease presentation. ESC Heart Fail 4: 365-370. [Crossref]
- Pieroni M, De Santis M, Zizzo G, Bosello S, Smaldone C et al. (2014) Recognizing and treating myocarditis in recent-onset systemic sclerosis heart disease: potential utility of immunosuppressive therapy in cardiac damage progression. Semin Arthritis Rheum 43: 526-535. [Crossref]
- Faludi R, Költő G, Bartos B, Csima G, Czirják L et al. (2014) Five-year follow-up of left ventricular diastolic function in systemic sclerosis patients: determinants of mortality and disease progression. Semin Arthritis Rheum 44: 220-227. [Crossref]
- Bissell LA, Yusof M, Yuzaiful M, Buch MH (2017) Primary myocardial disease in scleroderma: a comprehensive review of the literature to inform the UK Systemic Sclerosis Study Group cardiac working group. Rheumatology 56: 882-895. [Crossref]
- Kazzam E, Caidahl K, Hällgren R, Gustafsson R, Landelius J et al. (1991) Non-invasive assessment of systolic left ventricular function in systemic sclerosis. Eur Heart J 12: 151-156. [Crossref]
- Tyndall AJ, Bannert B, Vonk M, Airò P, Cozzi F et al. (2010) Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 69: 1809-1815. [Crossref]
- Tzelepis GE, Kelekis NL, Plastiras SC, Mitseas P, Economopoulos N et al. (2007) Pattern and distribution of myocardial fibrosis in systemic sclerosis: a delayed enhanced magnetic resonance imaging study. Arthritis Rheum 56: 3827-3836. [Crossref]
- Yiu KH, Schouffoer AA, Marsan NA, Ninaber MK, Stolk J et al. (2011) Left ventricular dysfunction assessed by speckle-tracking strain analysis in patients with systemic sclerosis: relationship to functional capacity and ventricular arrhythmias. Arthritis Rheum 63: 3969-3978. [Crossref]
- Ranque B, Bérezné A, Le-Guern V, Pagnoux C, Allanore Y et al. (2010) Myopathies related to systemic sclerosis: a case-control study of associated clinical and immunological features. Scand J Rheumatol 39: 498-505. [Crossref]
- Clemson BS, Miller WR, Luck JC, Feriss JA (1992) Acute myocarditis in fulminant systemic sclerosis. Chest 101: 872-874. [Crossref]
- Burt RK, Oliveira MC, Shah SJ, Moraes DA, Simoes B et al. (2013) Cardiac involvement and treatment-related mortality after non-myeloablative haemopoietic stem-cell transplantation with unselected autologous peripheral blood for patients with systemic sclerosis: a retrospective analysis. Lancet. 381: 1116-1124. [Crossref]
- Martens E, Lange P, Pohl T, Nickel T, Juchem G et al. (2012) Heart transplantation in a 36-year-old experiencing terminal heart failure caused by systemic sclerosis. Transplantation 94: e13-e15. [Crossref]
- Bogdanov I, Kazandjieva J, Darlenski R, Tsankov N (2018) Dermatomyositis: Current concepts. Clin Dermatol 36: 450-458. [Crossref]
- Callen JP, Wortmann RL (2006) Dermatomyositis. Clin Dermatol 24: 363-373. [Crossref]
- Dilaveris P, Pietri P, Tsiachris D, Gatzoulis K, Stefanadis C (2012) Inducible ventricular tachycardia due to dermatomyositis-related cardiomyopathy in the era of implantable cardioverter-defibrillator therapy. Circulation 125: 967-969. [Crossref]
- Lu Z, Guo‐chun W, Li M, Ning Z (2012) Cardiac involvement in adult polymyositis or dermatomyositis: a systematic review. Clin Cardiol 35: 685-691. [Crossref]
- Haupt HM, Hutchins GM (1982) The heart and cardiac conduction system in polymyositis-dermatomyositis: a clinicopathologic study of 16 autopsied patients. Am J Cardiol 50: 998-1006. [Crossref]
- Denbow CE, Lie JT, Tancredi RG, Bunch TW (1979) Cardiac involvement in polymyositis: a clinicopathologic study of 20 autopsied patients. Arthritis Rheum 22: 1088-1092. [Crossref]
- Finsterer J, Stöllberger C, Avanzini M, Rauschka H (2006) Restrictive cardiomyopathy in dermatomyositis. Scand J Rheumatol 35: 229-232. [Crossref]
- Erlacher P, Lercher A, Falkensammer J, Nassonov EL, Samsonov MI et al. (2001) Cardiac troponin and beta-type myosin heavy chain concentrations in patients with polymyositis or dermatomyositis. Clin Chim Acta 306: 27-33. [Crossref]
- Lundberg IE (2006) The heart in dermatomyositis and polymyositis. Rheumatology (Oxford) 45: iv18-iv21. [Crossref]
- Allanore Y, Vignaux O, Arnaud L, Puéchal X, Pavy S et al. (2006) Effects of corticosteroids and immunosuppressors on idiopathic inflammatory myopathy related myocarditis evaluated by magnetic resonance imaging. Ann Rheum Dis 65: 249-252. [Crossref]
- Ozkan Y (2016) Cardiac involvement in ankylosing spondylitis. J Clin Med Res 8: 427-430. [Crossref]
- Momeni M, Taylor N, Tehrani M (2011) Cardiopulmonary manifestations of ankylosing spondylitis. Int J Rheumato 2011: 728471 [Crossref]
- Papagoras C, Voulgari PV, Drosos AA (2013) Atherosclerosis and cardiovascular disease in the spondyloarthritides, particularly ankylosing spondylitis and psoriatic arthritis. Clin Exp Rheumatol 31: 612-620. [Crossref]
- O'Neill TW, Bresnihan B (1992) The heart in ankylosing spondylitis. Ann Rheum Dis 51: 705-706. [Crossref]
- O'Neill TW, King G, Graham IM, Molony J, Bresnihan B (1992) Echocardiographic abnormalities in ankylosing spondylitis. Ann Rheum Dis 51: 652-654. [Crossref]
- Lehtinen K (1993) Mortality and causes of death in 398 patients admitted to hospital with ankylosing spondylitis. Ann Rheum Dis 52: 174-176. [Crossref]
- Yildirir A, Aksoyek S, Calguneri M, Aytemir K, Kabakci G et al. (2000) QT dispersion as a predictor of arrhythmic events in patients with ankylosing spondylitis. Rheumatology (Oxford) 39: 875-879. [Crossref]
- Khan MA (1994) Ankylosing spondylitis. Clinical features. In: Klippel JH, Dieppe PA. Rheumatology. St. Louis: Mosby 25: 1-10.
- Gould BA, Turner J, Keeling DH, Hickling P, Marshall AJ (1992) Myocardial dysfunction in ankylosing spondylitis. Ann Rheum Dis 51: 227-232. [Crossref]
- Heeneman S, Daemen MJ (2007) Cardiovascular risks in spondyloarthritides. Curr Opin Rheumatol 19: 358-362. [Crossref]
- Bolten WW (2006) Problem of the atherothrombotic potential of non-steroidal anti-inflammatory drugs. Ann Rheum Dis 65: 7-13. [Crossref]
- Goodson NJ, Brookhart AM, Symmons DP, Silman AJ, Solomon DH (2009) Non-steroidal anti-inflammatory drug use does not appear to be associated with increased cardiovascular mortality in patients with inflammatory polyarthritis: results from a primary care-based inception cohort of patients. Ann Rheum Dis 68: 367-372. [Crossref]
- Bakland G, Gran JT, Nossent JC (2011) Increased mortality in ankylosing spondylitis is related to disease activity. Ann Rheum Dis 70: 1921-1925. [Crossref]
- Van der Heijde D, Sieper J, Maksymowych WP, Dougados M, Burgos-Vargas R, et al. (2011) 2010 Update of the international ASAS recommendations for the use of anti-TNF agents in patients with axial spondyloarthritis. Ann Rheum Dis 70: 905-908. [Crossref]
- Furst DE, Keystone EC, Braun J, Breedveld FC, Burmester GR, et al. (2012) Updated consensus statement on biological agents for the treatment of rheumatic diseases, 2011. Ann Rheum Dis 71: i2-i45. [Crossref]
- Barnabe C, Martin BJ, Ghali WA (2011) Systematic review and meta-analysis: anti-tumor necrosis factor a therapy and cardiovascular events in rheumatoid arthritis. Arthritis Care Res (Hoboken) 63: 522-529. [Crossref]
- Dixon WG, Watson KD, Lunt M, Hyrich KL, Silman AJ, et al. (2007) Reduction in the incidence of myocardial infarction in patients with rheumatoid arthritis who respond to anti-tumor necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Arthritis Rheum 56: 2905-2912. [Crossref]
- Daïen CI, Duny Y, Barnetche T, Daurès JP, Combe B, et al. (2012) Effect of TNF inhibitors on lipid profile in rheumatoid arthritis: a systematic review with meta-analysis. Ann Rheum Dis 71: 862-868. [Crossref]
- Zumla A, James DG (1996) Granulomatous infections: aetiology and classification. Clin Infect Dis 23: 146-158. [Crossref]
- James DG (2000) A clinicopathological classification of granulomatous disorders. Postgrad Med J 76: 457-465. [Crossref]
- Baughman RP, Lower EE, du Bois RM (2003) Sarcoidosis. Lancet 361: 1111-1118. [Crossref]
- Muchtar E, Blauwet LA, Gertz MA (2017) Restrictive cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res 121: 819-837. [Crossref]
- Lakdawala NK, Givertz MM (2010) Dilated cardiomyopathy with conduction disease and arrhythmia. Circulation 122: 527-534. [Crossref]
- Kim JS, Judson MA, Donnino R, Gold M, Cooper LT Jr et al. (2009) Cardiac sarcoidosis. Am Heart J 157: 9-21. [Crossref]
- Statement on sarcoidosis: joint statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee (1999) Am J Respir Crit Care Med 160: 736-755. [Crossref]
- Doughan AR, Williams BR (2006) Cardiac sarcoidosis. Heart 92: 282-288. [Crossref]
- Mantini N, Williams B Jr, Stewart J, Rubinsztain L, Kacharava A (2012) Cardiac sarcoid: a clinician’s review on how to approach the patient with cardiac sarcoid. Clin Cardiol 35: 410-415. [Crossref]
- Youssef G, Beanlands RS, Birnie DH, Nery PB (2011) Cardiac sarcoidosis: applications of imaging in diagnosis and directing treatment. Heart 97: 2078-2087. [Crossref]
- Ohira H, Tsujino I, Ishimaru S, Oyama N, Takei T et al. (2008) Myocardial imaging with F-fluoro-2-deoxyglucose positron emission tomography and magnetic resonance imaging in sarcoidosis. Eur J Nucl Med Mol Imaging 35: 933-941. [Crossref]
- Buckley O, Doyle L, Padera R, Lakdawala N, Dorbala S et al. (2010) Cardiomyopathy of uncertain etiology: complementary role of multimodality imaging with cardiac MRI and 18FDG PET. J Nucl Cardiol 17: 328-332. [Crossref]
- Yazaki Y, Isobe M, Hiroe M, Morimoto S, Hiramitsu S et al. (2001) Central Japan Heart Study Group. Prognostic determinants of long-term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol 88: 1006-1101. [Crossref]
- Comarmond C, Pagnoux C, Khellaf M, Cordier JF, Hamidou M et al. (2013) Eosinophilic granulomatosis with polyangiitis (Churg‐Strauss): Clinical characteristics and long‐term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum 65: 270-281. [Crossref]
- Kyaw H, Misra D, Mani MM, Park WJ, Shinnar M (2017) Unusual cardiac involvement in granulomatosis with polyangiitis manifesting as acute congestive heart failure. Anatol J Cardiol 18: 158-160. [Crossref]
- Kubaisi B, Samra KA, Foster CS (2016) Granulomatosis with polyangiitis (Wegener's disease): An updated review of ocular disease manifestations Intractable Rare Dis Res 5: 61-69. [Crossref]
- John R, Herzenberg AM (2009) Vasculitis affecting the kidney. Semin Diagn Pathol 26: 89-102. [Crossref]
- Mahr AD, Neogi T, Merkel PA (2006) Epidemiology of Wegener's granulomatosis: Lessons from descriptive studies and analyses of genetic and environmental risk determimants. Clin Exp Rheumatol 24: S82-91. [Crossref]
- Walsh M, Flossmann O, Berden A, Westman K, Hoglund P et al. (2012) Risk factors for relapse of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 64: 542-548. [Crossref]
- Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L et al. (2011) The five-factor score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine 90: 19-27. [Crossref]
- Lynch JP, Tazelaar H (2011) Wegener granulomatosis (granulomatosis with polyangiitis): Evolving concepts in treatment. Semin Respir Crit Care Med 32: 274-297. [Crossref]
- Knockaert DC (2007) Cardiac involvement in systemic inflammatory diseases. Eur Heart J 28: 1797-1804. [Crossref]
- Lim HE, Lee YH, Ahn JC (2007) Wegener’s granulomatosis with progressive conduction disturbances and atrial fibrillation. Heart 93: 777. [Crossref]
- Marques N, Mimoso J, Bohorquez R, Lázaro M, Brito H et al. (2008) Unstable angina as initial manifestation of Wegener granulomatosis: case report. Rev Port Cardiol 27: 1469-1477. [Crossref]
- Mukhopadhyay S, Hensley RG, Tazelaar HD (2010) Cardiac involvement in Wegener granulomatosis diagnosed at autopsy. Cardiovasc Pathol 19: 312-315. [Crossref]
- Grant SC, Levy RD, Venning MC, Ward C, Brooks NH (1994) Wegener's granulomatosis and the heart. Br Heart J 71: 82-86. [Crossref]
- To A, De Zoysa J, Christiansen JP (2007) Cardiomyopathy associated with Wegener’s granulomatosis. Heart 93: 984. [Crossref]
- Dupuy M, Marcheix B, Grunenwald E, Dickson Z, Cron C et al. (2009) Mitral regurgitation associated with Wegener's granulomatosis. Ann Cardiol Angeiol (Paris) 58: 180-182. [Crossref]
- Brihaye B, Aouba A, Pagnoux C, Vignaux O, Le Hello C et al. (2008) Rituximab reversed cardiac involvement of Wegener's granulomatosis: magnetic resonance imaging assessment. Presse Med 37: 412-425. [Crossref]
- Fauci AS, Haynes BF, Katz P, Wolff SM (1983) Wegener’s granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med 98: 76-85. [Crossref]
- Weidhase A, Grone HJ, Unterberg C, Schuff-Werner P, Wiegand V (1990) Severe granulomatous giant cell myocarditis in Wegener’s granulomatosis. Klin Wochenschr 68: 880-885. [Crossref]
- Korzets Z, Chen B, Levi A, Pomeranz A, Bernheim J (1991) Non dilated congestive cardiomyopathy—a fatal sequelae of Wegener’s granulomatosis. J Nephrol 1: 61-64.
- Day JD, Ellison KE, Schnittger I, Perlroth MG (1996) Wegener’s granulomatosis presenting as dilated cardiomyopathy. West J Med 165: 64-66. [Crossref]
- Delevaux I, Hoen B, Selton-Suty C, Canton P (1997) Relapsing congestive cardiomyopathy in Wegener’s granulomatosis. Mayo Clin Proc 72: 848-850. [Crossref]
- 123To A, De Zoysa J, Christiansen JP (2007) Cardiomyopathy associated with Wegener’s granulomatosis. Heart 93: 984. [Crossref]
- Sarlon G, Durant C, Grandgeorge Y, Bernit E, Veit V et al. (2010) Cardiac involvement in Wegener’s granulomatosis: report of four cases and review of the literature. Rev Méd Interne 31: 135-139. [Crossref]
- Lynch JP III, White E, Tazelaar H, Langford CA (2004) Wegener’s granulomatosis: evolving concepts in treatment. Semin Respir Crit Care Med 25: 491-521. [Crossref]
- Kallenberg CG (2007) Antineutrophil cytoplasmic autoantibody associated small-vessel vasculitis. Curr Opin Rheumatol 19: 17-24. [Crossref]
- Kallenberg CG (2008) Pathogenesis of PR3-ANCA associated vasculitis. J Autoimmun 30: 29-36. [Crossref]
- Gomez-Puerta JA, Bosch X (2009) Anti-neutrophil cytoplasmic antibody pathogenesis in small-vessel vasculitis: an update. Am J Pathol 175: 1790-1798. [Crossref]
- Dijstelbloem HM, Scheepers RH, Oost WW, Stegeman CA, Van Der Pol WL et al. (1999) Fcgamma receptor polymorphisms in Wegener’s granulomatosis: risk factors for disease relapse. Arthritis Rheum 42: 1823-1827. [Crossref]
- Savige JA, Chang L, Cook L, Burdon J, Daskalakis M et al. (1995) Alpha 1-antitrypsin deficiency and anti-proteinase 3 antibodies in anti-neutrophil cytoplasmic antibody (ANCA)-associated systemic vasculitis. Clin Exp Immunol 100: 194-197. [Crossref]
- Borgmann S, Endisch G, Urban S, Sitter T, Fricke H (2001) A linkage disequilibrium between genes at the serine protease inhibitor gene cluster on chromosome 14q32.1 is associated with Wegener’s granulomatosis. Clin Immunol 98: 244-248. [Crossref]
- Leavitt RY, Fauci AS, Bloch DA, Michel BA, Hunder GG et al. (1990) The American College of Rheumatology 1990 criteria for the classification of Wegener's granulomatosis. Arthritis Rheum 33: 1101-1107. [Crossref]
- To A, De Zoysa J, Christiansen JP (2007) Cardiomyopathy associated with Wegener's granulomatosis. Heart 93: 984. [Crossref]
- Walton EW (1958) Giant-cell granuloma of the respiratory tract (Wegener’s granulomatosis). BMJ 2: 265-270. [Crossref]
- Hoffman GS (1994) Wegener’s granulomatosis: the path travelled since 1931. Medicine (Baltimore) 73: 325-329. [Crossref]
- Hollander D, Manning RT (1967) The use of alkylating agents in the treatment of Wegener’s granulomatosis. Ann Intern Med 67: 393-398. [Crossref]
- Fauci AS, Wolff SM (1973) Wegener’s granulomatosis: studies in eighteen patients and a review of the literature. Medicine (Baltimore) 52: 535-561. [Crossref]
- Guillevin L, Cordier JF, Lhote F, Cohen P, Jarrousse B et al. (1997) A prospective, multicenter, randomized trial comparing steroids and pulse cyclophosphamide versus steroids and oral cyclophosphamidein the treatment of generalized Wegener’s granulomatosis. Arthritis Rheum 40: 2187-2198. [Crossref]
- Pagnoux C, Hogan SL, Chin H, Jennette JC, Falk RJ et al. (2008) Predictors of treatment resistance and relapse in antineutrophil cytoplasmic antibodyassociated small-vessel vasculitis: comparison of two independent cohorts. Arthritis Rheum 58: 2908-2918. [Crossref]
- Talar-Williams C, Hijazi YM, Walther MM, Linehan WM, Hallahan CW et al. (1996) Cyclophosphamide-induced cystitis and bladder cancer in patients with Wegener granulomatosis. Ann Intern Med 124: 477-484. [Crossref]
- Charlier C, Henegar C, Launay O, Pagnoux C, Berezne A et al. (2009) Risk factors for major infections in Wegener granulomatosis: analysis of 113 patients. Ann Rheum Dis 68: 658-663. [Crossref]
- Faurschou M, Sorensen IJ, Mellemkjaer L, Loft AG, Thomsen BS et al. (2008) Malignancies in Wegener’s granulomatosis: incidence and relation to cyclophosphamide therapy in a cohort of 293 patients. J Rheumatol 35: 100-105. [Crossref]
- Suranagi VV, Malur PR, Bannur HB (2009) Cardiac sarcoidosis causing sudden death. Indian Journal of Pathology and Microbiology 52: 566-567. [Crossref]
- Cooper LT Jr, Berry GJ, Shabetai R (1997) Idiopathic giant-cell myocarditis– natural history and treatment. Multicenter Giant Cell Myocarditis Study Group Investigators. N Engl J Med 336: 1860-1866. [Crossref]
- Kandolin R, Lehtonen J, Salmenkivi K, Räisänen-Sokolowski A, Lommi J et al. (2013) Diagnosis, treatment, and outcome of giant-cell myocarditis in the era of combined immunosuppression. Circ Heart Fail 6: 15-22. [Crossref]
- Izumi T, Kodama M, Shibata A (1991) Experimental giant cell myocarditis induced by cardiac myosin immunization. Eur Heart J 12: 166-168. [Crossref]
- Kittleson MM, Minhas KM, Irizarry RA, Ye SQ, Edness G et al. (2005) Gene expression in giant cell myocarditis: altered expression of immune response genes. Int J Cardiol 102: 333-340. [Crossref]
- Cooper LT Jr, Hare JM, Tazelaar HD, Edwards WD, Starling RC et al. (2008) Giant Cell Myocarditis Treatment Trial Investigators. Usefulness of immunosuppression for giant cell myocarditis. Am J Cardiol 102: 1535-1539. [Crossref]
- Okura Y, Dec GW, Hare JM, Kodama M, Berry GJ et al. (2003) A clinical and histopathologic comparison of cardiac sarcoidosis and idiopathic giant cell myocarditis. J Am Coll Cardiol 41: 322-328. [Crossref]
- Cooper LT (2000) Giant cell myocarditis: diagnosis and treatment. Herz 25: 291-298. [Crossref]
- Lagana SM, Parwani AV, Nichols LC (2010) Cardiac sarcoidosis: a pathology focused review. Arch Pathol Lab Med 134: 1039-1046. [Crossref]
- Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J et al. (2013) Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 34: 2636-2648. [Crossref]
- Xu J, Brooks EG (2016) Giant cell myocarditis: a brief review. Arch Pathol Lab Med 140: 1429-1434. [Crossref]
- Suarez-Barrientos A, Wong J, Bell A, Lyster H, Karagiannis G et al. (2015) Usefulness of rabbit anti-thymocyte globulin in patients with giant cell myocarditis. Am J Cardiol 116: 447-445. [Crossref]
- Elamm C, Al-Kindi S, Oliveira G (2016) Heart transplant in giant cell myocarditis: a contemporary analysis of the UNOS database. J Card Fail 22: S124.
- Murray LK, González-Costello J, Jonas SN, Sims DB, Morrison KA et al. (2012) Ventricular assist device support as a bridge to heart transplantation in patients with giant cell myocarditis. Eur J Heart Fail 14: 312-318. [Crossref]
- Bennett MK, Gilotra NA, Harrington C, Rao S, Dunn JM et al. (2013) Evaluation of the role of endomyocardial biopsy in 851 patients with unexplained heart failure from 2000-2009. Circ Heart Fail 6: 676-684. [Crossref]
- Kanai-Yoshizawa S, Sugiyama Kato T, Mancini D, Marboe CC (2013) Hypersensitivity myocarditis and outcome after heart transplantation. J Heart Lung Transplant 32: 553-559. [Crossref]
- Takkenberg JJ, Czer LS, Fishbein MC, Luthringer DJ, Quartel AW et al. (2004) Eosinophilic myocarditis in patients awaiting heart transplantation. Crit Care Med 32: 714-721. [Crossref]
- Haas SJ, Hill R, Krum H, Liew D, Tonkin A et al. (2007) Clozapine-associated myocarditis: a review of 116 cases of suspected myocarditis associated with the use of clozapine in Australia during 1993-2003. Drug Saf. 30: 47-57. [Crossref]
- Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M et al. (2007) The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. Circulation 116: 2216-2233. [Crossref]
- Eckart RE, Love SS, Atwood JE, Arness MK, Cassimatis DC et al. (2004). Incidence and follow-up of inflammatory cardiac complications after smallpox vaccination. J Am Coll Cardiol 44: 201-205. [Crossref]
- Engler RJ, Nelson MR, Collins LC Jr, et al. (2015) A prospective study of the incidence of myocarditis/pericarditis and new onset cardiac symptoms following smallpox and influenza vaccination. PLoS One 10: e0118283. [Crossref]
- Taubert KA, Rowley AH, Shulman ST (1991) Nationwide survey of Kawasaki disease and acute rheumatic fever. J Pediatr 119: 279-282. [Crossref]
- Gordon JB, Kahn AM, Burns JC (2009) When children with Kawasaki disease grow up: myocardial and vascular complications in adulthood. J Am Coll Cardiol 54: 1911-1920. [Crossref]
- Etsuda H, Takase B, Ohsuzu F, Ishihara M, Kurita A (2006) Poor prognostic cardiac sequelae of possible Kawasaki disease mimicking dilated cardiomyopathy: the importance of extensive and serial cardiac evaluation and the significance of thromboembolic mechanisms. Anadolu Kardiyol Derg 6: 275-278. [Crossref]
- Liu AM, Ghazizadeh M, Onouchi Z, Asano G (1999) Ultrastructural characteristics of myocardial and coronary microvascular lesions in Kawasaki disease. Microvasc Res 58: 10-27. [Crossref]
- Kato H, Sugimura T, Akagi T, et al. (1996) Long-term consequences of Kawasaki disease. A 10- to 21-year follow-up study of 594 patients. Circulation 94:1379-1385. [Crossref]
- Burns JC, Glode MP (2004) Kawasaki syndrome. Lancet 364: 533-544. [Crossref]
- Senzaki H (2008) Long-term outcome of Kawasaki disease. Circulation 118: 2763-2772. [Crossref]
- Tsuda E, Matsuo M, Naito H, Noguchi T, Nonogi H et al. (2007) Clinical features in adults with coronary arterial lesions caused by presumed Kawasaki disease. Cardiol Young 17: 84-89. [Crossref]
- Tatara K, Kusakawa S, Itoh K, et al. (1989) Long-term prognosis of Kawasaki disease patients with coronary artery obstruction. Heart Vessels 5: 47-51. [Crossref]
- Shiraishi I, Onouchi Z, Hayano T, Hamaoka K, Kiyosawa N (1991) Asymptomatic myocardial infarction in Kawasaki disease: long-term prognosis. Pediatr Cardiol 12: 78-82. [Crossref]
- Tsuda E, Kamiya T, Ono Y, Kimura K, Kurosaki K, Echigo S (2005) Incidence of stenotic lesions predicted by acute phase changes in coronary arterial diameter during Kawasaki disease. Pediatr Cardiol 26: 73-79. [Crossref]
- Muneuchi J, Joo K, Morihana E, Mizushima A (2008) Detectable Silent Calcification in a Regressed Coronary Artery Aneurysm of a Young Adult with a History of Kawasaki Disease. Pediatr Cardiol 29: 195-197. [Crossref]
- Arnold R, Goebel B, Ulmer HE, Gorenflo M, Poerner TC (2007) An exercise tissue Doppler and strain rate imaging study of diastolic myocardial dysfunction after Kawasaki syndrome in childhood. Cardiol Young 17: 478-486. [Crossref]
- Tsuda E, Arakaki Y, Shimizu T, et al. (2005) Changes in causes of sudden deaths by decade in patients with coronary arterial lesions due to Kawasaki disease. Cardiol Young 15: 481-488. [Crossref]
- Nagasawa H, Arakaki Y, Yamada O, Nakajima T, Kamiya T (1996) Longitudinal observations of left ventricular end-diastolic dimension in children using echocardiography. Pediatr Cardiol 17: 169-674. [Crossref]
- Kurotobi S, Kawakami N, Shimizu K, et al. (2005) Brain Natriuretic Peptide as a Hormonal Marker of Ventricular Diastolic Dysfunction in Children with Kawasaki Disease. Pediatr Cardiol. 2005 [Crossref]
- Takeuchi D, Saji T, Takatsuki S, Fujiwara M (2006) Abnormal tissue doppler images are associated with elevated plasma brain natriuretic peptide and increased oxidative stress in acute Kawasaki disease. Circ J 71: 357-362. [Crossref]
- Yagi S, Tsuda E, Shimizu W, et al. (2005) Two adults requiring implantable defibrillators because of ventricular tachycardia and left ventricular dysfunction caused by presumed Kawasaki disease. Circ J 69: 870-874. [Crossref]
- Lappé JM, Pelfrey CM, Tang WW (2008) Recent insights into the role of autoimmunity in idiopathic dilated cardiomyopathy. J Card Fail 14: 521-530. [Crossref]
- Pankuweit S, Portig I, Maisch B (2002) Pathophysiology of cardiac inflammation: molecular mechanisms. Herz 27: 669-676. [Crossref]
- Kyvik KO, Green A, Beck-Nielsen H (1995) Concordance rates of insulin dependent diabetes mellitus: a population-based study of young Danish twins. BMJ 311: 913-917. [Crossref]
- Willer CJ, Dyment DA, Risch NJ, Sadovnick AD, Ebers GC (2003) Twin concordance and sibling recurrence rates in multiple sclerosis. Proc Natl Acad Sci U S A 100: 12877-12882. [Crossref]
- Limas CJ, Iakovis P, Anyfantakis A, Kroupis C, Cokkinos DV (2004) Familial clustering of autoimmune diseases in patients with dilated cardiomyopathy. Am J Cardiol 93: 1189-1191. [Crossref]
- Carlquist JF, Menlove RL, Murray MB, O'Connell JB, Anderson JL (1991) HLA class II (DR and DQ) antigen associations in idiopathic dilated cardiomyopathy. Validation study and meta-analysis of published HLA association studies. Circulation 83: 515-522. [Crossref]
- McKenna CJ, Codd MB, McCann HA, Sugrue DD (1997) Idiopathic dilated cardiomyopathy: familial prevalence and HLA distribution. Heart 77:549-552. [Crossref]
- Konstadoulakis MM, Kroumbouzou H, Tsiamis E, Trikas A, Toutouzas P (1993) Clinical significance of antibodies against tropomyosin, actin and myosin in patients with dilated cardiomyopathy. J Clin Lab Immunol 40: 61-67. [Crossref]
- Stork S, Boivin V, Horf R, Hein L, Lohse MJ et al. (2006) Stimulating autoantibodies directed against the cardiac beta1-adrenergic receptor predict increased mortality in idiopathic cardiomyopathy. Am Heart J 152: 697-704. [Crossref]
- Baba A, Yoshikawa T, Fukuda Y, Sugiyama T, Shimada M et al. (2004) Autoantibodies against M2-muscarinic acetylcholine receptors: new upstream targets in atrial fibrillation in patients with dilated cardiomyopathy. Eur Heart J 25: 1108-1115. [Crossref]
- Lauer B, Schannwell M, Kuhl U, Strauer BE, Schultheiss HP (2000) Anti-myosin autoantibodies are associated with deterioration of systolic and diastolic left ventricular function in patients with chronic myocarditis. J Am Coll Cardiol 35: 11-18. [Crossref]
- Muller J, Wallukat G, Dandel M, Bieda H, Brandes K et al. (2000) Immunoglobulin adsorption in patients with idiopathic dilated cardiomyopathy. Circulation 101:385-391. [Crossref]
- Galvin JE, Hemric ME, Kosanke SD, Factor SM, Quinn A et al. (2002) Induction of myocarditis and valvulitis in lewis rats by different epitopes of cardiac myosin and its implications in rheumatic carditis. Am J Pathol 160: 297-306. [Crossref]
- Dangas G, Konstadoulakis MM, Epstein SE, Stefanadis CI, Kymionis GD et al. (2000) Prevalence of autoantibodies against contractile proteins in coronary artery disease and their clinical implications. Am J Cardiol 85: 870-872. [Crossref]
- Li Y, Heuser JS, Cunningham LC, Kosanke SD, Cunningham MW (2006) Mimicry and antibodymediated cell signaling in autoimmune myocarditis. J Immunol 2006;177: 8234-8240. [Crossref]
- Staudt Y, Mobini R, Fu M, Felix SB, Kuhn JP et al. (2003). Beta1-adrenoceptor antibodies induce apoptosis in adult isolated cardiomyocytes. Eur J Pharmacol 466: 1-6. [Crossref]
- Ferrari I, Levin MJ, Wallukat G, Elies R, Lebesgue D et al. (1995) Molecular mimicry between the immunodominant ribosomal protein P0 of Trypanosoma cruzi and a functional epitope on the human beta 1-adrenergic receptor. J Exp Med 182: 59-65. [Crossref]
- Mobini R, Staudt A, Felix SB, Baumann G, Wallukat G et al. (2003) Hemodynamic improvement and removal of autoantibodies against beta1-adrenergic receptor by immunoadsorption therapy in dilated cardiomyopathy. J Autoimmun 20: 345-350. [Crossref]
- Jahns R, Boivin V, Siegmund C, Inselmann G, Lohse MJ et al (1999) Autoantibodies activating human beta1-adrenergic receptors are associated with reduced cardiac function in chronic heart failure. Circulation 99: 649-654. [Crossref]
- Miao GB, Liu JC, Liu MB, Wu JL, Zhang G et al. (2006) Autoantibody against beta1adrenergic receptor and left ventricular remodeling changes in response to metoprolol treatment. Eur J Clin Invest 36: 614-620. [Crossref]
- Horwich TB, Patel J, MacLellan WR, Fonarow GC (2003) Cardiac troponin I is associated with impaired hemodynamics, progressive left ventricular dysfunction, and increased mortality rates in advanced heart failure. Circulation 108: 833-838. [Crossref]
- Goser S, Andrassy M, Buss SJ, Leuschner F, Volz CH et al. (2006) Cardiac troponin I but not cardiac troponin T induces severe autoimmune inflammation in the myocardium. Circulation 114: 1693-702. [Crossref]
- Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A et al. (2003) Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med 9: 1477-1483. [Crossref]
- Shmilovich H, Danon A, Binah O, Roth A, Chen G et al. (2007) Autoantibodies to cardiac troponin I in patients with idiopathic dilated and ischemic cardiomyopathy. Int J Cardiol 117: 198-203. [Crossref]
- Baba A, Yoshikawa T, Ogawa S (2002) Autoantibodies produced against sarcolemmal Na-K-ATPase: possible upstream targets of arrhythmias and sudden death in patients with dilated J Am Coll Cardiol 40: 1153-1159. [Crossref]
- Kuhl U, Noutsias M, Seeberg B, Schultheiss HP (1996) Immunohistological evidence for a chronic intramyocardial inflammatory process in dilated cardiomyopathy. Heart 75: 295-300. [Crossref]
- Noutsias M, Pauschinger M, Schultheiss H, Kh U (2002) Phenotypic characterization of infiltrates in dilated cardiomyopathy—diagnostic significance of T-lymphocytes and macrophages in inflammatory cardiomyopathy. Med Sci Monit 8: CR478-487. [Crossref]
- Omerovic E, Bollano E, Andersson B, Kujacic V, Schulze W et al. (2000) Induction of cardiomyopathy in severe combined immunodeficiency mice by transfer of lymphocytes from patients with idiopathic dilated cardiomyopathy. Autoimmunity 32: 271-280. [Crossref]
- Lunardi C, Tiso M, Borgato L, Nanni L, Millo R et al. (1998) Chronic parvovirus B19 infection induces the production of anti-virus antibodies with autoantigen binding properties. Eur J Immunol 28: 936-948. [Crossref]
- Wucherpfennig KW, Strominger JL (1995) Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 80: 695-705. [Crossref]
- Lapierre P, Beland K, Alvarez F (2007) Pathogenesis of autoimmune hepatitis: from break of tolerance to immune-mediated hepatocyte apoptosis. Transl Res 149: 107-113. [Crossref]
- Hughes RA, Allen D, Makowska A, Gregson NA (2006) Pathogenesis of chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst 11: 30-46. [Crossref]
- Urowitz MB, Gladman D, Ibañez D, Bae SC, Sanchez-Guerrero J et al. (2010) Systemic Lupus International Collaborating Clinics. Atherosclerotic vascular events in a multinational inception cohort of systemic lupus erythematosus. Arthritis Care Res (Hoboken) 62: 881-887. [Crossref]
- Tlustochowicz W, Piotrowicz R, Cwetsch A, Raczka A, Kramarz E et al. (1995) 24-h ECG monitoring in patients with rheumatoid arthritis. Eur Heart J 16: 848-851. [Crossref]
- Chapelon-Abric C, de Zuttere D, Duhaut P, Veyssier P, Wechsler B et al. (2004) Cardiac sarcoidosis: a retrospective study of 41 cases. Medicine 83: 315-334. [Crossref]
- Forsblad-d’Elia H, Wallberg H, Klingberg E, Carlsten H, Bergfeldt L (2013) Cardiac conduction system abnormalities in ankylosing spondylitis: a cross-sectional study. BMC Musculoskelet Disord 14: 237. [Crossref]
- Touma Z, Harvey P, Gladman D, Sabapathy A, Urowitz MB (2013) lupus Patients Have a High Prevalence Of Abnormalities On Resting Electrocardiogram That Are Associated With Increased Risk For Cardiovascular Events.: 623. Arthritis & Rheumatism 65: S269
- Bacani AK, Crowson CS, Roger VL, Gabriel SE, Matteson EL (2015) Increased incidence of atrial fibrillation in patients with rheumatoid arthritis. Biomed Res Int 2015: 809514 [Crossref]
- Bourre‐Tessier J, Urowitz MB, Clarke AE, Bernatsky S, Krantz MJ et al. (2015) Electrocardiographic findings in systemic lupus erythematosus: data from an international inception cohort. Arthritis Care Res 67: 128-135. [Crossref]
- Chauhan K, Ackerman MJ, Crowson CS, Matteson EL, Gabriel SE (2015) Population-based study of QT interval prolongation in patients with rheumatoid arthritis. Clin Exp Rheumatol 33: 84-89. [Crossref]
- Geraldino-Pardilla L, Gartshteyn Y, Piña P, Cerrone M, Giles JT et al. (2016) ECG non-specific ST-T and QTc abnormalities in patients with systemic lupus erythematosus compared with rheumatoid arthritis. Lupus Sci Med 3: e000168. [Crossref]
- Masooleh IS, Zayeni H, Haji-Abbasi A, Azarpira M, Hadian A et al. (2016) Cardiac involvement in rheumatoid arthritis: A cross-sectional study in Iran. Indian Heart J 68: 332-335. [Crossref]
- Al Rayes H, Harvey PJ, Gladman DD, Su J, Sabapathy A et al. (2017) Prevalence and associated factors of resting electrocardiogram abnormalities among systemic lupus erythematosus patients without cardiovascular disease. Arthritis Res Ther 19: 31. [Crossref]
- El-Monem SM, Ali AY, Hashaad NI, Bendary AM, El-Aziz HA (2019) Association of rheumatoid arthritis disease activity, severity with electrocardiographic findings, and carotid artery atherosclerosis. Egyptian Rheumatology and Rehabilitation 46: 11.
- Di Franco M, Paradiso M, Mammarella A, Paoletti V, Labbadia G et al. (2000) Diastolic function abnormalities in rheumatoid arthritis. Evaluation by echo Doppler transmitral flow and pulmonary venous flow: relation with duration of disease. Ann Rheum Dis 59: 227-229. [Crossref]
- Guedes C, Bianchi‐Fior P, Cormier B, Barthelemy B, Rat AC et al. (2001) Cardiac manifestations of rheumatoid arthritis: a case–control transesophageal echocardiography study in 30 patients. Arthritis Rheum 45: 129-135. [Crossref]
- Hameed S, Malik LM, Shafi S, Azeem S, Shahzad A (2007) Echocardiographic evaluation of patients with systemic lupus erythematosus. Pakistan Journal of Medical Sciences 23: 497.
- Allam NT, Darweesh HE, Hamadnallah N, Ashour ZA (2013) Evaluation of left ventricular myocardial function in Egyptian patients with systemic lupus erythematosus: Tissue Doppler study and its relation to disease activity. Egyptian Rheumatologist 35: 217-223.
- Bakhoum SW, El Basel M, Alnaggar AR, Hamdy MS, Hussein H (2015) Assessment of left ventricular function in systemic lupus erythematosus patients by speckle tracking echocardiography: Relation to circulating endothelial progenitor cells. Egyptian Rheumatologist 37: S33-S41.
- Klingberg E, Sveälv BG, Täng MS, Bech-Hanssen O, Forsblad-d'Elia H et al. (2015) Aortic regurgitation is common in ankylosing spondylitis: time for routine echocardiography evaluation? Am J Med 128: 1244-1250. [Crossref]
- Yazici A, Cosan F, Yilmazer B, Cefle A (2015) AB0334 Cardiac Echocardiography Findings of Patients with Rheumatoid Arthritis. Ann Rheum Dis 74: 1005.
- Mathew D, Bastian C (2017) Clinical Study of Cardiovascular Involvement in Patients with Connective Tissue Diseases. Open J Rheumatol Autoimmune Dis 7: 178.
- Hannawi S, Naeem K, Hannawi H, Al Salmi I (2018) Echocardiographic Abnormalities in Rheumatoid Arthritis Patients Compared to Age, Sex and Traditional Cardiovascular Risk Factors Matched Controls. Arthritis & Rheumatology 70: 540.
- Mohammed MA, Rady SA, El-Mokadem MO, Fadda SM (2018) Echocardiographic findings in systemic lupus erythematosus and its relation to disease activity and damage index. The Egyptian Rheumatologist 40: 173-178.
- Mohamed AAA, Hammam N, EL Zohri MH, Gheita TA (2019) Cardiac manifestations in systemic lupus erythematosus: clinical correlates of subclinical echocardiographic features. BioMed Res Int 2019: 2437105.
- Caforio AL, Adler Y, Agostini C, Allanore Y, Anastasakis A et al. (2017) Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Disease. Eur Heart J 38: 2649-2662. [Crossref]
- Chow PC1, Ho MH, Lee TL, Lau YL, Cheung YF (2007) Relation of arterial stiffness to left ventricular structure and function in adolescents and young adults with pediatric-onset systemic lupus erythematosus. J Rheumatol 34: 1345-1352. [Crossref]
- Corrao S, Argano C, Pistone G, Messina S, Calvo L et al. (2015) Rheumatoid arthritis affects left ventricular mass: systematic review and meta-analysis. Eur J Intern Med 26: 259-267. [Crossref]
- Sasson Z, Rasooly Y, Chow CW, Marshall S, Urowitz MB (1992) Impairment of left ventricular diastolic function in systemic lupus erythematosus. Am J Cardiol 69: 1629-1634. [Crossref]
- Roldan CA, Tolstrup K, Macias L, Qualls CR, Maynard D et al. (2015) Libman-Sacks endocarditis: detection, characterization, and clinical correlates by three-dimensional transesophageal echocardiography. J Am Soc Echocardiogr 28: 770-779. [Crossref]
- Rudski LG1, Lai WW, Afilalo J, Hua L, Handschumacher MD et al. (2010) Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr 23: 685-713. [Crossref]
- Galie` N, Humbert M, Vachiery JL, Gibbs S, Lang I et al. (2016) 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 37: 67-119. [Crossref]
- Hachulla AL, Launay D, Gaxotte V, de Groote P, Lamblin N et al. (2009) Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 68: 1878-1884. [Crossref]
- Allanore Y, Meune C (2010) Primary myocardial involvement in systemic sclerosis: evidence for a microvascular origin. Clin Exp Rheum 28: S48-S53. [Crossref]
- Leone O, Veinot JP, Angelini A, Baandrup UT, Basso C et al. (2012) 2011 consensus statement on endomyocardial biopsy from the Association for European Cardiovascular Pathology and the Society for Cardiovascular Pathology. Cardiovasc Pathol 21: 245-274. [Crossref]
- Ardehali H, Howard DL, Hariri A, Qasim A, Hare JM et al. (2005) A positive endomyocardial biopsy result for sarcoid is associated with poor prognosis in patients with initially unexplained cardiomyopathy. Am Heart J 150: 459-463. [Crossref]
- Austin BA, Duffy B, Tan C, Rodriguez ER, Starling RC et al. (2009) Comparison of functional status, electrocardiographic, and echocardiographic parameters to mortality in endomyocardial-biopsy proven cardiac amyloidosis. Am J Cardiol 103: 1429-1433. [Crossref]
- Baccouche H, Mahrholdt H, Meinhardt G, Merher R, Voehringer M et al. (2009) Diagnostic synergy of non-invasive cardiovascular magnetic resonance and invasive endomyocardial biopsy in troponin-positive patients without coronary artery disease. Eur Heart J 30: 2869-2879. [Crossref]
- Thurberg BL, Fallon JT, Mitchell R, Aretz T, Gordon RE et al. (2009) Cardiac microvascular pathology in Fabry disease: evaluation of endomyocardial biopsies before and after enzyme replacement therapy. Circulation 119: 2561-256. [Crossref]
- Dinarello CA, Simon A, van der Meer JW (2012) Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 11: 633-652. [Crossref]
- Cho JH, Feldman M (2015) Heterogeneity of autoimmune diseases: pathophysiologic insights from genetics and implications for new therapies. Nat Med 21: 730-738. [Crossref]
- Rose NR (2016). Viral myocarditis. Curr Opin Rheumatol 28: 383-389. [Crossref]
- Jensen LD, Marchant DJ (2016) Emerging pharmacologic targets and treatments for myocarditis. Pharmacol Ther 161: 40-51. [Crossref]
- Ekström K, Lehtonen J, Kandolin R, Räisänen‐Sokolowski A, Salmenkivi K et al. (2016) Long-term outcome and its predictors in giant cell myocarditis. Eur J Heart Fail 18: 14521458. [Crossref]
- Frustaci A, Russo MA, Chimenti C (2009) Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: the TIMIC study. Eur Heart J 30: 1995-2002. [Crossref]
- Mason JW, O'connell JB, Herskowitz A, Rose NR, McManus BM et al. (1995) A clinical trial of immunosuppressive therapy for myocarditis. The Myocarditis Treatment Trial Investigators. N Engl J Med 333: 269-275. [Crossref]
- Menghini VV, Savcenko V, Olson LJ, Tazelaar HD, Dec GW et al. (1999) Combined immunosuppression for the treatment of idiopathic giant cell myocarditis. Mayo Clin Proc 74: 1221-1226. [Crossref]
- Bulkley BH, Roberts WC (1975) The heart in systemic lupus erythematosus and the changes induced in it by corticosteroid therapy. A study of 36 necropsy patients. Am J Med 58: 243-264. [Crossref]
- Herda LR, Trimpert C, Nauke U, Landsberger M, Hummel A (2010) Effects of immunoadsorption and subsequent immunoglobulin G substitution on cardiopulmonary exercise capacity in patients with dilated cardiomyopathy. Am Heart J 159: 809-816. [Crossref]
- Knebel F, Böhm M, Staudt A, Borges AC, Tepper M et al. (2004) Reduction of morbidity by immunoadsorption therapy in patients with dilated cardiomyopathy. Int J Cardiol 97: 517-520. [Crossref]
- Staudt A, Staudt Y, Hummel A, Empen K, Dörr M et al. (2006) Effects of immunoadsorption on the nt-BNP and nt-ANP plasma levels of patients suffering from dilated cardiomyopathy. Ther Apher Dial 10: 42-48. [Crossref]
- Kishimoto C, Shioji K, Hashimoto T, Nonogi H, Lee JD et al. (2014) Therapy with immunoglobulin in patients with acute myocarditis and cardiomyopathy: analysis of leukocyte balance. Heart Vessel 29: 336-342. [Crossref]
- McNamara DM, Holubkov R, Starling RC, Dec GW, Loh E et al. (2001) Controlled trial of intravenous immune globulin in recent onset dilated cardiomyopathy. Circulation 103: 2254-2259. [Crossref]
- Drucker NA, Colan SD, Lewis AB, Beiser AS, Wessel DL et al. (1994) Gamma-globulin treatment of acute myocarditis in the paediatric population. Circulation 89: 252-257. [Crossref]