Abstract
Arrhythmogenic right ventricular dysplasia (ARVD) is a serious heart muscle disease, often inherited. It is implicated as a leading cause of exercise-related sudden death primarily in young adults and athletes. Developing a precise understanding of its prevalence, clinical course and manifestation would improve early detection and help institute prompt and appropriate therapeutic management. Thus, this review article aggregates current research-based evidence on ARVD. The intention is to advance the current understanding and status of ARVD to improve diagnosis and survival rates. Particularly, this article reviews clinical description, classification, epidemiology, clinical manifestation, prognosis, risk factors, pathophysiology, etiology, differential diagnosis and clinical management of ARVD.
Key words
ventricular dysplasia, cardiomyopathy, ventricular arrhythmias
Introduction
Historical context
The earliest historical suggestion of arrhythmogenic right ventricular dysplasia (ARVD), also known as arrhythmogenic right ventricular cardiomyopathy (ARVC), was in 1736 in the book “De Motu Cordis et Aneurysmatibus” (On the Motion of the Heart and on Aneurysms) by Giovanni Maria Lancisi, the Pope’s physician and Professor of Anatomy in Rome. The book details the recurrence of a non-specific cardiac disease in four generations in which family members presented with palpitations, heart failure, dilation, right ventricular aneurysm and sudden death [1]. Dalla-Volta et al. in 1961 published an article on “auricularization” of RV pressure curve, where the right ventricle was behaving like an atrium, providing additional suspicion of a cardiac disorder due to global right ventricular dysfunction [2].
However, it was not until 1977 that Fontaine et al. [3] first proposed the term ARVD and its seminal description as a clinical entity. The study described six patients with sustained ventricular tachycardia not explained by any overt heart disease and non-responsive to antiarrhythmic drugs. There were also reports of localized right ventricular involvement later supported with evidence that ARVD can progress and diffuse to RV and LV leading to bi-ventricular heart failure [4,5]. In 1982, Marcus et al. [6] published the first clinical profile of ARVD, describing 24 cases presenting with advanced disease presenting with enlarged LV resulting from fibrofatty infiltration of the RV free wall and genetic association. Later in 1984, Thiene et al. [7] made the first description of electrocardiographic (ECG) abnormalities related to ARVD including the epsilon wave, a diagnostic criterion found in 30% of ARVD patients. Ten years later (in 1994), the first gene locus ARVD1 was identified at chromosome 14q23 followed in 1996 by a detailed description of a pathological genetic profile of ARVD involving the left ventricle [8].
In 1996, ARVD was formally recognized as a form of cardiomyopathy by the revised publication of the World Health Organization (WHO) and International Society and Federation of Cardiology (ISFC) task force on definition and classification of cardiomyopathies [9]. The task force included ARVD as one of the five morphological and functional phenotype of cardiomyopathies. The other four were dilated cardiomyopathy, hypertrophic cardiomyopathy, restrictive cardiomyopathy and unclassified cardiomyopathy.
The position statement from the European Society of Cardiology (ESC) working group on myocardial and pericardial diseases proposed a definition of ARVD based on histological findings. They define ARVD by “the presence of right ventricular dysfunction (global or regional) with or without left ventricular disease in the presence of histological evidence for the disease and/or electrocardiographic abnormalities” [10]. The ESC further define histological findings as progressive pathological replacement of RV myocardium by adipose and fibrous tissues usually confined to RV inflow, outflow and apex, collectively referred to as the ‘triangle of dysplasia’ (Figure 1), which lead to morphological and functional abnormalities in the RV myocardium. The pathological changes may also occur in the LV myocardium mimicking or leading to a dilated cardiomyopathy phenotype [11]. Subsequent studies have provided similar ARVD definitions based on pathological and clinical findings. Pathologically, they define ARVD as a heart muscle disorder often familial marked by pathological replacement of RV myocardium by fibrofatty tissues and clinically by the presence of life-threatening ventricular arrhythmias originating from the RV that potentially leads to cardiac death mostly in young adults and athletes or ultimately may be a subtle cause of congestive heart failure [5,8,12].
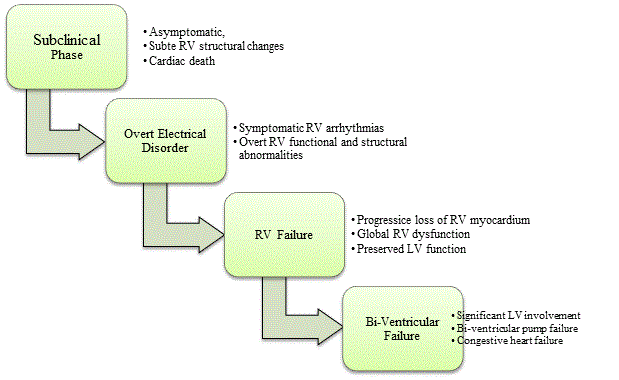
Figure 1: Clinical Course of Arrhythmogenic Right Ventricular Dysplasia
The clinical course of ARVD has five main stages: asymptomatic subclinical phase, symptomatic phase characterized with overt electrical disorder leading to RV failure and finally bi-ventricular failure. Source: Corrado et al. 2000, p,3 [20]; Thiene et al., 2007, p.3 [1]
Epidemiology
The exact prevalence of ARVD in the general population remains unclear due to a lack of large-scale or population-based epidemiological studies. However, studies using small nationally defined samples provide rough estimates of the prevalence of ARVD. In German, estimated ARVD prevalence is 1 in 5,000 people [13] and in Italy, Veneto Region, the prevalence varies between 1 in 2,000 to 5,000 people [14]. Other reported ARVD prevalence vary between 1 in 1,000 people [15], 1 in 2,500 to 5,000 people [1], and 1 in 5,000 [10,16, 17]. Based on several research findings, Basso et al. [18] state the estimated prevalence of ARVD ranges between 1 in 1,000 to 1 in 5,000 people.
Clinical presentation
Growing clinical and pathological evidence establishes ARVD as a disorder characterized by progressive loss of the myocardium and its replacement by fibrofatty tissues [1,19,20]. Clinical studies with long-term follow up demonstrate that overtime the right ventricle becomes more diffusely involved in disease progression [19,21,22]. Ultimately, ARVD progresses with significant involvement of the left ventricular, leading to bi-ventricular congestive heart failure [20]. The clinical course of ARVD is a function of both progressive ventricular dysfunction and functional (electrical) abnormality. There is research consensus that the clinical course consists of four main clinocopathologic phases, namely: subclinical phase, overt electrical disorder phase, RV failure phase and finally bi-ventricular (LV and RV) failure phase [19,21]. Figure 1 illustrates the four clinical phases alongside their characteristic morphological and functional symptoms.
Subclinical phase
The subclinical or concealed phase is asymptomatic, characterized by subtle RV structural changes in the presence or absence of minor ventricular arrhythmias [1]. Sudden cardiac death maybe the first and only manifestation of the disorder mostly in young adults during intense physical exercise or sporting competitions [19]. Figure 2 illustrates typical morphological ventricular features of asymptomatic young adult who died suddenly of ARVD. In Figure 2 (A) is four-chamber view of the heart revealing myocardial fatty replacement in RV fee wall and in the translucent infundibulum while in Figure 2 (B) is a histological view of the same heart confirming fatty replacement is confined only to RV wall but absent in the LV free wall and inter-ventricular septum.
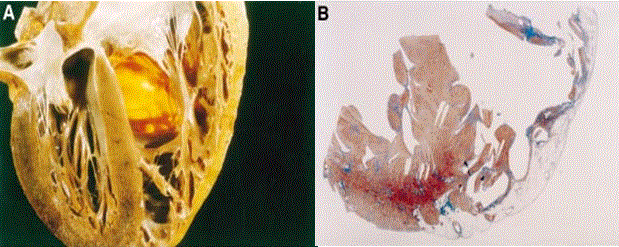
Figure 2: Morphological Features of 25-year old Man Who Died Suddenly of ARVD
Morphological features of a 25-yr old man who died suddenly of ARVD. (A) 4-chqamber view of the heart specimen revealing fibrofatty replacement of RV free wall. (B) Panoramic histological view of the same heart revealing myocardial atrophy in the RV. Source: Corrado, Basso & Thiene, 2000, p.589 [12]
Dsc2: desmocollin-2; Dsg2: desmoglein-2; Dsp: desmoplakin; Pkg: Plakoglobin; Pkp2: plakophilin-2, PM: Plasma Membrane
The subclinical phase progresses into the overt electrical disorder phase, which may present with palpitation and syncope. The most frequent clinical manifestation is symptomatic ventricular arrhythmias originating from RV usually triggered by intense exercise [12,19]. Ventricular arrhythmias maybe isolated premature beats, sustained tachycardia with left bundle branch block (LBB) morphology or ventricular fibrillation potentially causing cardiac death. During ventricular tachycardia, QRS axis and morphology suggest the site of origin. Left bundle branch block with inferior axis suggest arrhythmias originate from RV outflow tract while Left bundle branch block with supervisor axis suggests arrhythmias originate RV inferior wall [1].
RV failure
The third clinical phase of ARVD is marked by RV failure caused by a progressive loss of the RV myocardium. The loss provokes global RV dysfunction but with relatively preserved LV function. Global RV dysfunction may account for severe pump failure in this clinical phase [19,22].
Bi-ventricular failure
Bi-ventricular failure represent the end-stage of the progressive clinical course of ARVD. This stage is characterized by biventricular failure due to significant involvement of the LV and interventricular septum. Bi-ventricular failure may lead to congestive heart disease mimicking dilated bi-ventricular cardiomyopathy [19]. In the setting of atrial fibrillation, endocavitary mural thrombosis may occur in the atrial appendages or within aneurysms to account for thromboembolic events leading to severe contractile dysfunction often requiring cardiac transplantation [1].
Cardiologists should suspect ARVD in young adults presenting with syncope, ventricular tachycardia, cardiac arrest or adult patients with congestive heart failure [23,24]. Besides these signs and symptoms, clinical three main clinical presentation of ARVD include arrhythmias, sudden death and heart failure.
Ventricular arrhythmias ranging from isolated premature beats to tachycardia remains the primary suspicion for ARVD patients in clinical practice [24-27]. In the subclinical phase, many AVRD probands having a history of syncope may have subtle RV structural changes with or without arrhythmias, may remain undiagnosed because of the absence of other suggestive features [12,28]. Ventricular arrhythmias may cause palpitations, syncope or sudden cardiac death [29]. In ARVD patients with ventricular tachycardia, arrhythmias usually manifest with LBB morphology indicating the point of origination is the RV [5,12]. Some patients present with multiple ventricular tachycardia morphologies due to the disorder producing multiple arrhythmogenic foci [25].
Sudden cardiac death (SCD) is an important clinical manifestation of ARVD. Although an uncommon cause of SCD, in the U.S., ARVD accounts for an estimated 5% of SCD in patients aged 65 years or below [30] and between 3% and 4% in exercise-related SCDs in young athletes [31]. In Veneto region Italy, ARVD is the most frequently encountered cause of death in clinical practice in individuals aged 35 years or below usually associated with intense exercise [6,32]. Estimated annual mortality rate of ARVD ranges from 1% with pharmacotherapy to 3% if left untreated. The suspected mechanism of ARVD-related SCD is acceleration of ventricular tachycardia degenerating into ventricular fibrillation or the degradation in functional and structural RV performance [29].
Patients with ARVD may develop isolated right heart failure or bi-ventricular heart failure, which usually manifest in the third and fourth decades of life [13]. ARVD is one of very few myocardial disorders in the absence of pulmonary hypertension that causes right heart failure. The mechanism of right ventricular failure is dilation, thinning of wall and continued loss of contractile function due to myocardial atrophy [29]. In ARVD, right heart failure manifests four to eight years after the occurrence of an ECG-indicated right bundle branch block (RBBB) [30]. Whereas left heart failure is uncommon in ARVD, the disorder can involve LV and induce mild changes in LV function. If ARVD causes heart failure, there is a likelihood of misdiagnosis of dilated cardiomyopathy of idiopathic or viral origin [13]. In most cases, LV dysfunction in ARVD represents biventricular dysplasia, which should be distinguished from biventricular fibrotic myocarditis [33]. Distinction is important since ARVD patients a more likely to present with drug-resistance arrhythmias and sudden cardiac death and thus require other therapeutic options [29].
The primary objective of clinical management of ARVD patients is the prevention of life threatening ventricular arrhythmias [17]. This objective however is undermined by the lack of prospective and controlled studies evaluating clinical risk markers that can assist in predicting the appearance of life-threatening ventricular arrhythmias [17,20]. Several studies [1,12,19,22] have already established that in the subclinical phase, sudden cardiac death may be the first and only manifestation of ARVD mostly in asymptomatic young adults and athletes. They indicates both diagnosed and suspected ARVD patients are at risk of sudden cardiac death even in the absence of symptoms or lethal ventricular arrhythmias. A challenging clinical dilemma is to whether to consider primary prevention, prophylactic therapy on asymptomatic ARVD patients or those with minor symptoms diagnosed during routine family screening, and whether to offer secondary prevention to ARVD patients presenting with malignant ventricular arrhythmias. Moreover, ARVD is a progressive myocardial disease with an increasing risk of death overtime [20].
The risk factors for ARVD-related sudden cardiac death reported from retrospective studies analyzing clinicopathologic factors and fatal ARVD incidents suggest young age, intense exercise or competitive sporting activities, malignant family history, extensive right ventricular disease with reduced LV ejection fraction, and LV ventricular involvement, syncope and ventricular tachycardia are significant risk factors [23,31,34,34]. There are reports of at least 80% of ARVD cases diagnosed before the age of 40 years [23] and intense exercise accounting for up to 4% of deaths in young adults [31] and significantly lower survival free of ventricular tachycardia or ventricular fibrillation and heart failure [35]. An estimated 20% to 30% ARVD patients have a family history of ARVD or sudden cardiac death [34] while a recent study using non-invasive tests including electrocardiography and echocardiography observe evidence of ARVD in between 30% and 50% of family members [7,36,37]. Further, there is a 50% probability that ARVD mutations in the proband will be passed to the offspring, since most genetic mutations responsible for ARVD have an autosomal dominant pattern of inheritance with variable penetrance [38].
Morphological hallmark of ARVD is the progressive loss and replacement of myocardium in the RV free wall by fibrofatty tissue [12]. Only the subendocardial layers remain relatively preserved, in which myocardium is interspersed with fibrosis [39]. The replacement is progressive, beginning at the epicardium then extending to the endocardium to become transmural, which accounts for dilatations and aneurysms in the triangle of dysplasia reported in about 50% of the autopsy case series [5,7,8]. The fibrofatty replacement of the RV free wall disrupts electrical conduction within the ventricles causing delays (late potential or epsilon waves) and re-entrant excitations, which are mechanisms that explain ventricular arrhythmias [1]. However, emerging research evidence suggests fibrofatty replacement of the RV free wall is an inadequate morphological hallmark for clinical identification of ARVD. Some amounts of intramyocardial fat have been observed in the RV anterolateral and apical areas in the normal heart increasing with age and body size [41]. Routine autopsy may also overlook fibrofatty replacement because of the lack of a clear distinction between normal and pathological fatty infiltration [20]. Further, fat interspersed with cardiomyocytes have been observed in hearts of a significant number of normal adults [41, 42].
Corrado et al. [12,20] suggested two variants of fatty replacement of the myocardium in ARVD: the purely fatty (adipose) and the fibrofatty variants. The purely fatty variant is characterized by partial or complete fatty replacement of the myocardium without wall thinning, defined as RV free wall thickness of 4-5 mm. The fatty replacement predominantly affects the apex and infundibulum, and the lack fibrotic tissues and inflammatory infiltrates suggests the left ventricle and intraventricular septum are preserved [7,8,40]. In half of the cases of the purely fatty variant, there are reports of myocardial generation and death in the absence of other concomitant heart disease [20].
The fibrofatty variant was the original morphological definition of ARVD. This variant is characterized by fatty infiltration associated with significant replacement by fibrotic tissues, thinning of RV free wall (wall thickness < 3mm), aneurysmal dilatation and inflammatory infiltrates [12,20]. Half of autopsy case series observe fibrofatty replacement in RV apical, inflow and outflow regions, “the triangle of dysplasia” but rarely involves the left ventricle and ventricular septum [7,12]. Corrado et al. [20] propose three basic mechanisms underlying the loss of myocardium and its replacement with fibrofatty tissues. (a) Apoptosis (programmed cell death). (b) Inflammatory cardiac conditions manifesting with a range of clinical conditions – acute myocarditis to fibrous healing – that may involve both LV and RV leading to congestive heart failure mimicking dilated cardiomyopathy. (c) Myocardial dystrophy unrelated to myocarditis suggesting inherited atrophy.
Etiology of ARVD is predominantly genetic but other conditions such as congenital abnormalities and viral or inflammatory myocarditis affect RV or facilitate genetic expression of the disorder [29,43].
Inherited genetic mutation has been frequently cited as the dominant etiology of ARVD indicated in many studies that define ARVD as a heterogeneous hereditary myocardial disorder usually with an autosomal dominant pattern of inheritance [34,44-46]. The suspected genetic etiopathogenesis of ARVD may be explained by a purely structural theory illustrated in Figure 4. Abnormalities in the myocardial cell-to-cell connection through contacts referred to as intercalated discs. Portion of the discs could also have abnormalities referred to as the desmosome. The desmosome disrupts the normal structure and function of desmosomal proteins leading to intercalated discs pulling apart [34,38].
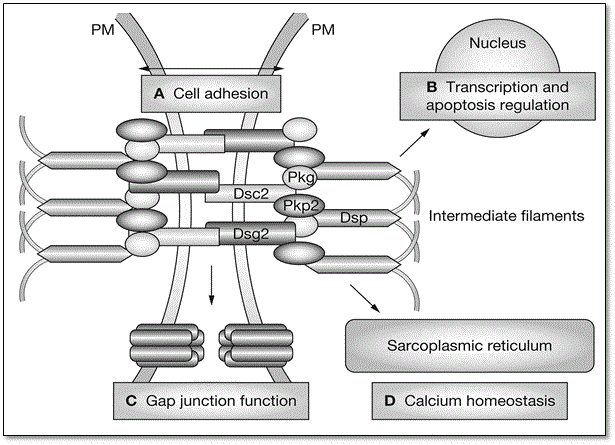
Figure 3: Structure of Intercalated Discs and Role of Desmosome
Mutations in various cardiac desmosomal genes and their four main roles involved in etiopathogenesis of ARVD. (A) cell-adhesion supporting structural stability; (B) regulation of transcription and apoptosis; (C) regulate gap junction; and (D) regulate calcium ion homeostasis. Adapted from Awad et al., 2008, p.13 [38].
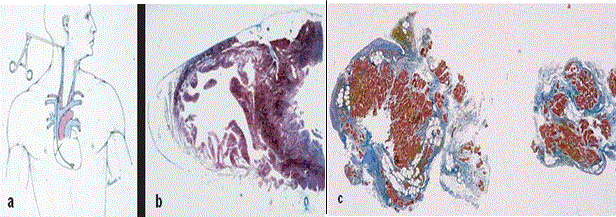
Figure 4: Endomyocardial Biopsy Characterization of RV Free Wall in ARVD Patients
In vitro tissue characterization of endomyocardial biopsy. (a) transvenous jugular approach of the bioptome; (b) cross-sectional area of the heart revealing the septum is spared; (c) fibrofatty replacement of two bioptic samples. Source: Thiene et al., 2007, p.6 [1]
Figure 3 illustrates mutations in various cardiac desmosomal genes with important roles in the etiopathogenesis of ARVD. The desmosomal cardiac genes play four important roles in the normal structure and function of myocardium. They support structural stability (cell-to-cell adhesion) (A), regulate transcription in genes involved in adipogenesis, apoptosis and in the maintenance of normal electrical conductivity (B), regulate gap junction (C) and regulation of calcium ion (Ca+) in the extracellular fluid (D) [38]. The most affected myocardial cells are in the RV but as ARVD progress, RV becomes involved, and in rare cases, the LV might be an early or predominant site of ARVD [43]. The disruption of the normal structure of the myocardium may lead to life-threatening arrhythmias, RV dysfunction and sudden cardiac death [38].
Recent studies on electron microscopy have supported the purely structural theory postulating the loss of myocyte adhesion causes necrosis and fibrosis to explain genetic pathogenesis of ARVD [47,48]. The two studies report ultrastructural abnormalities of the desmosome (focal scar) linked with desmosomal gene mutations. Focal scar would lead to the characteristic ARVD-related RV arrhythmias with environmental factors such as intense exercise or inflammation cause by viral infection could stimulate the pulling apart of the desmosome and exacerbate disease progression [38]. The RV is at a higher risk of the loss of myocyte adhesion due to thinner wall and normal dilatory response to exercise. However, the structural theory fails to explain preserved LV myocardium and late onset of the disorder in some individuals having desmosomal gene mutation [38].
Currently, five desmosomal genes have been identified that correlate with early onset of clinical symptoms and life-threatening ventricular arrhythmias in ARVD: plakophilin-2, desmoglein-2, desmoscollin-2, desmoplakin and plakoglobin [27,34,38]. In two studies investigating desmosomal genetic mutations and the distribution of gene defects in desmosome proteins recruiting a large series of ARVD patients, 26.7% [49] and 58% [50] had positive gene defects in desmosome proteins with plakophilin-2 being the most prevalent genetic mutation accounting for 73% [49] and 78.4% [50] as summarized in Table 1.
Table 1: Desmosomal Genetic Mutation and Distribution in ARVD Patients
Positive Gene Defects in ARVD Patients |
Desmosomal Gene Defects |
Study 1 |
Study 2 |
Total Patients (N) |
198 |
175 |
Positive Genetic Defects |
26.7% |
58% |
Distribution of Desmosomal Gene Defects |
Plakophilin-2 |
73% |
78.4% |
Desmoglein-2 |
9.6% |
13.5% |
Desmoscollin-2 |
5.8% |
3.6% |
Desmoplakin |
7.7% |
2.7% |
Plakoglobin |
3.8% |
0.9% |
Source: 1Xu et al. [49]; 2Kapplinger et al. [50].
Compared to other inherited cardiomyopathic phenotypes with a single causative genetic mutation, the genetic etiology of ARVD is more complex. Patients with ARVD may have more than one genetic defect in the same gene, a condition referred to as compound heterozygosity, or more than one genetic defect in a commentary gene, referred to as digenic heterozygosity [38]. Based on heterogeneous genetic etiologies, ARVD has been classified into eleven (11) genetic variants. Table 2 provides a summary of eleven identified genetic variants of ARVD.
Table 2: Genetic Variants of ARVD
Variant |
Transmission |
Gene |
Characteristic |
ARVD1 |
AD |
TGFβ3 |
Progressive RV myocardium loss
Cytokine stimulation of fibrosis & cell adhesion modulation |
ARVD2 |
AD |
Cardiac ryanodine receptor |
4 mutations in different families linked to catecholaminergic polymorphic (CP) VT
Whether CP and ARVD2 are different mutations of the same gene is unclear. |
ARVD3 |
AD |
-- |
-- |
ARVD4 |
AD |
-- |
Local LV involvement with LBBB in some patients |
ARVD5 |
AD |
-- |
Seen in few ARVD cases |
ARVD6 |
AD |
-- |
Early onset and high penetrance |
ARVD7 |
AD |
-- |
Linked to myofibrillar myopathy |
ARVD8 |
AD
AR |
Desmoplakin |
Linked with palmoplantar keratoderma and woolly hair |
ARVD9 |
AR |
Plakophilin-2 |
Presents with arrhythmia earlier than other ARVD mutations |
ARVD10 |
AD |
Desmoglein-2 |
Component of the desmosome gene |
ARVD11 |
AD |
Desmoglein-2 |
Mutation results in frameshift and premature truncation |
AD: Autosomal Dominant; AR: Autosomal Recessive; CP: Catecholaminergic Polymorphic; VT: Ventricular Tachycardia
Source: Awad et al., 2008, p.277 [38]
Genetic etiologies have been reported as dominant in ARVD patients but only between 30% and 50% of ARVD patients have one of the five identified genes suggesting the involvement of other not-yet identified causative genes [38]. Awad et al. [38] reviewed studies on novel non-desmosomal genes, which identify genetic mutations in cardiac ryanodine receptor 2 (RYR2), which has been associated with pathogenesis of ARVD2 genetic variant. Mutations in RYR2 is also linked to catecholaminergic polymorphic ventricular, a disorder resembling ARVD but without RV abnormality as reported in 12 individuals [51]. Linkage analysis have reported a relationship between chromosome 1q42.1-q43 and ARVD phenotype identified in RYR2 mutations in four families [52]. However, the role of RYR2 mutations remains ambiguous because catecholaminergic polymorphic ventricular tachycardia is a separate clinical disorder with unclear clinical criteria for diagnosis. It is also not established whether catecholaminergic polymorphic and ARVD2 are disorder of different mutation of the same gene [38].
Regulatory mutations in TGFβ is another non-desmosomal gene mutation suspected to cause ARVD. Gene defect in TGFβ is associated with a tendency for increased production of extracellular matrix and adipogenesis [53]. Continuing studies on transgenic mice tested with available genetic mutations to provide better understanding of etiopathogenesis mechanism of genetic ARVD with the potential for therapeutic implications [54,55].
Whereas genetic etiologies predominate literature on the causes of ARVD, other non-genetic causes and theories have been proposed. Mostly, incomplete penetrance and variable phenotypic expression in ARVD suggest the role of non-genetic or environmental influences [18,25,43]. The high prevalence of ARVD in highly athletic individuals or those participating in competitive sporting activities, who have a five-fold increased risk strengthening the suggestion of etiopathogenic role of environmental factors [56,57]. Congenital abnormalities involving the right ventricle and cardiac disorders such as viral or inflammatory myocarditis have been suggested as non-genetic etiologies of ARVD. Four theories have also been proposed over time to explain the cause of ARVD or as environmental conditions facilitating genetic expression of ARVD: (a) disontogenetic theory [8,58]; (b) degenerative theory [8,59]; (c) inflammatory theory [7,33,60,61]; (d) apoptotic theory [62,63]; and (e) myocyte trans-differentiation theory [64].
The disontogenetic theory is historical and associated ARVD to “patchment RV” referred at the time as Uhl’s anomaly. It proposed ARVD is congenital hypoplasia of the myocardium of the right ventricle presenting in infancy as congestive heart disease [8,58]. The degenerative theory postulated ARVD results from the death of heart cells caused by inherited ultrastructural or metabolic abnormalities [59]. Inflammatory theory proposes that ARVD results from previous myocarditis explained by inflammatory infiltrates observed in specimen from ARVD patients [60.61]. Apoptotic theory suggest ARVD results from apoptosis explained by the presence of cysteine protein required for apoptosis observed in diseases RV myocardium of six ARVD patients but absent in normal patients [62,63]. Finally, the trans-differentiation theory proposed that ARVD results from myocardial cells changing from muscle to fatty tissues underpinned by the finding that one ARVD patient has transitional cells interfacing myocardium and fatty tissues expressing desmin, a characteristic of muscle tissues and vimentin, found only in adipocytes [64].
Morphological and functional abnormalities of the RV myocardium in ARVD became apparent after the publication of the first clinical profile in 1982 based on 24 patient cases with advanced disease [6]. However, since then, ARVD has proved difficult to diagnose especially if the patient has mild or no discernible structural or functional changes to the RV myocardium. Bomma et al. [65] re-evaluated 65 patients diagnosed with ARVD based on MRI findings of intramyocardial fat or RV wall thinning from an outside institution and found none of the patients had ARVD, suggesting a high frequency of misdiagnosis especially diagnosis based purely on MRI wall thinning and fibrofatty replacement.
Diagnosis challenges and the lack of a gold standard or single test to diagnose ARVD informed the formation of the Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology (ESC) and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology (ISFC). The ESC/ISFC Task Force developed the initial standardized guidelines for the diagnosis of ARVD in 1994 [66] and later revised them in 1996 [9]. Table 3 summarizes the revised Task Force criteria for the diagnosis of ARVD. The standardized criteria streamlines and simplifies diagnosis but uncertainty persists because of the lack of quantified guidelines in many criteria and the lack of reports on outcomes of evaluation of large series of newly diagnosed ARVD patients using task force criteria [67,68].
Table 3: Task Force Criteria for ARVD Diagnosis
Criteria |
Major |
Minor |
Global/regional dysfunction & structural alterations* |
Severe dilation & reduction or RVEF with no/mild LV impairment |
Mild global RV dilation or EF reduction with normal LV. |
Localized RV aneurysms |
Regional RV hypokinesia |
Severe segmental RV dilation |
Mild segmental RV dilation |
Tissue characterization of walls |
Fibrofatty replacement of myocardium on endomyocardial biopsy. |
-- |
Repolarization abnormalities |
-- |
Inverted T waves in right precordial leads (V1 and V3) – people aged >12 years in absence of RBBB |
Depolarization/ Conduction Abnormalities |
Epsilon T waves or localized prolongation > 110 ms of the QRS complex in right precordial leads (V1 to V3) |
Late potentials (signal average ECG) |
Arrhythmias |
-- |
LBBB type VT (sustained/ non sustained) – ECG, Holter, exercise testing |
-- |
Frequent ventricular extrasystoles (>1,000/24 h) (Holter) |
Family History |
Familial disease confirmed at necropsy or surgery |
Familial history of premature sudden death <35 years due to suspected RV dysplasia. |
-- |
Familial history – clinical diagnosis based on present criteria |
RVEF: Right Ventricular Ejection Fraction; EF: Ejection Fraction; RBBB: Right Bundle Branch Block; LBBB: Left Bundle Branch Block; VT: Ventricular Tachycardia; ECG: Electrocardiography
* Detected by echocardiography, angiography, magnetic resonance imaging or radionuclide scintigraphy
Source: Gemayel et al. [29]; Corrado et al. [12,20]
The ESC/ISFC Task Force diagnostic criteria emphasizes on in vivo diagnosis, whose aim is to demonstrate alterations in the RV myocardium morphology and function. The alterations include abnormalities in depolarization and repolarization, arrhythmias of the LBBB morphology, fibrofatty substitution of myocardial tissue. The criteria describe myocardial changes detected by echocardiography, angiography, cardiac magnetic resonance imaging and/or radionuclide scintigraphy as well as other diagnostic criteria such as family history of the disorder [1].
Diagnosis of ARVD based on the Task Force criteria relies on the presence of major and minor clinical findings from structural, histologic, electrocardiographic, arrhythmic, and genetic factors. Confirmation of the diagnosis of ARVD requires the presence of two major criteria, one major and two minor criteria or four minor criteria from different groups. These diagnostic criteria represent a useful clinical approach that is specific but may lack sensitivity ultimately requiring molecular genetic testing upon which clinical diagnosis will be based [12,67].
Based on the Task Force Revised 1996 diagnostic criteria for ARVD, techniques for differential diagnosis focus on genetics, depolarization or repolarization abnormalities, arrhythmias and ventricular structural alterations or tissue characterization.
Genetic testing is helpful for determining whether an individual suspected to have ARVD actually has the disorder and identifying asymptomatic relatives having inherited ARVD-associated gene defects [37]. Genetic testing for a specific mutation is recommended when diagnosis of ARVD has been established in the proband to assess possible risk of the disease in close relatives [69]. Mutation specific tests determine whether close relatives (brothers, sisters and offspring) have the same causal gene defect suggesting they are at risk of ARVD requiring periodical evaluation to determine when the disease will manifest over time. Although the lack of gene defect in close family members suggest unlikeliness of disease development, the proband may have unidentified secondary gene defects that could also cause the disease to propagate to close family members [37].
Genetic testing has also been used in autopsy analysis to determine cause of sudden cardiac death in young adults suspected to have ARVD but diagnosis was inconclusive [70,71]. Genetic autopsy testing usually is done on blood samples whereby for a positive genetic test result, family screening of close relatives is recommended. Genetic tests also tests other causes of sudden cardiac death such as long QT syndrome if there is no discernible cause of death [70]. Genetic testing for ARVD has also been applied to prenatal diagnosis to inform the decision for ARVD parents to conceive a child [72]. In severe and refractory ARVD, ARVD parents may opt for in-vitro fertilization and the embryos tested for a specific inherited gene mutation before implantation to mother. Some ARVD parents may opt for prenatal diagnosis using amniocentesis at the start of pregnancy to determine fetal genetic status and inform the decision to keep or terminate the pregnancy if ethical and legal issues permit [37,72].
The complexity of genetic etiology of ARVD complicates the efficacy of genetic testing. Although the most common ARVD gene leading to clinical manifestation of ARVD is the plakophilin-2 gene, many ARVD patients have compound heterozygosity (more than one mutation in the same gene) or digenic heterozygosity (more than one mutation in a complementary gene) [38]. The plakophilin-2 gene thus may require compound or digenic heterozygosity to manifest clinical symptoms of ARVD, which challenges the accurate determination of the causative gene defect by genetic testing [37]. Defective ARVD causative desmosomal gene has only been identified in 1 in 400 individuals yet in a Finish population-prevalence study for ARVD desmosomal mutation, 1 in 200 health individuals have desmosomal gene mutations for ARVD [73] and 6% of healthy individuals of Asian descent have an abnormal plakophilin-2 gene [50,74].
Genetic testing for ARVD also has challenges in result interpretation due to multiple reports of 48% of individuals diagnosed with familial ARVD having multiple mutations in the same gene [49, 75-78]. These individuals usually present with more severe signs and symptoms. Since not all ARVD-associated genes have not been identified, determination of the causative defective gene may be problematic. Family members with an unknown defective mutation in many cases may have gene-negative findings. They should be considered for intermittent examinations to determine that they are not at-risk of ARVD [37]. Finally, the cost of genetic testing is high and prohibitive for many patients and/or families. An average cost of US$5,400 in the U.S. is high, which is exacerbated by some laboratories not accepting commercial insurance including Medicare or Medicaid [37].
Endomyocardial biopsy presents a major criterion for the diagnosis of ARVD through in vivo characterization of RV free wall with fibrofatty substitution of the myocardium [1]. Figure 4 shows RV free wall tissue characterization by endomyocardial biopsy. Figure 4(a) shows transvenous jugular approach of the bioptome; (b) cross-section of the heart revealing septum is spared suggesting biopsy at the level of RV free wall; and (c) fibrofatty substitution in two bioptic samples.
Confirmatory diagnosis of ARVD usually required histological assessment of transmural fibrofatty substitution of the myocardium at necropsy through either surgery or endomyocardial biopsy. However, diagnosis of ARVD by endomyocardial biopsy is challenging because of the segmental nature of the disease and low sensitivity because biopsy involves the interventricular septum, which is rarely involved in fibrofatty substitution [20,29,66].
The task force diagnostic criteria of ARVD recommend electrocardiography (abbreviated as ECG) for assessing changes in repolarization or depolarization and conduction abnormalities [9]. The recommendation is based on previous studies that demonstrated ECG recordings detect up to 90% of depolarization or repolarization abnormalities in ARVD patients [12]. Table 4 shows Task Force ECG criteria for diagnosis of familial (inherited) ARVD in the context of confirmed ARVD in a close relative (proband) [9,67].
Table 4: Major/Minor ECG Recordings for Suggestive Diagnosis of ARVD
Diagnosis Target |
Findings |
Suggestive ECG Recordings |
Repolarization Abnormalities |
Major |
Inverted T-waves in right precordial leads (V1,V2,V3) in individuals > 14 years in the absence of complete RBBB QRS ≥ 120 ms |
Minor |
Inverted T-waves in leads V1 and V2 in individuals > 14 years in the absence of complete RBBB or in V5-V6.
Inverted T-waves in leads V1-V4 in individuals > 14 years in the presence of complete RBBB |
Depolarization Abnormalities |
Major |
Epsilon wave in the right precordial leads (V1-V3) |
Minor |
Late potentials in Signal Averaged-ECG ≥ 1 of 3 parameters in the absence of QRS duration ≥ 110 ms on standard ECG |
Source: Marcus et al. 2010, pp.1535-1536 [67].
The most frequently encountered repolarization abnormality in clinical practice is T-wave inversion in precordial leads V1-V3 examining the RV. Figure 5 shows ECG recordings of a 19-year old football player who suddenly died from ARVD during competitive sporting activity. T-wave abnormality range between V1-V4 and isolated premature ventricular beats with LBBB morphology [12].
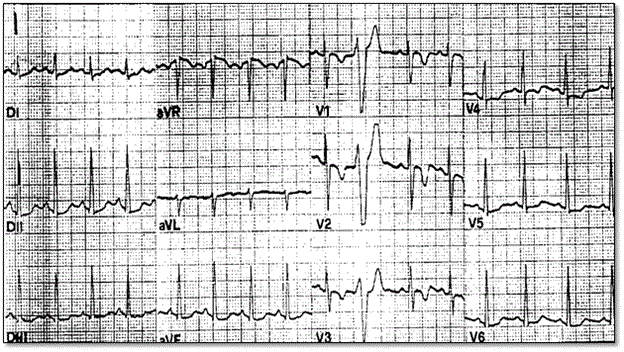
Figure 5: Typical 12-Lead ECG Recordings of T-wave Inversion in ARVD Patients
12-lead ECG obtained from participation screening of a 19-yr old footballer who died from ARVD during a competitive game. ECG abnormalities revealed were inverted T-waves (V1-V40 and isolated premature ventricular beats with left branch block morphology. Source: Corrado et al. 2000, p.589 [12].
Corrado et al. [12] links T-wave inversion with ST segment elevation <0.1 mV. The repolarization abnormalities are non-specific and considered minor diagnostic criteria because of a normal variant detected in females or children 12 years or younger, or occurs secondary to RBBB either isolated or in the context of congenital heart disease causing RV overload. The wide spectrum of ECG abnormalities suggesting delayed RV activation includes complete/incomplete RBBB, prolonged right precordial QRS duration and post-excitation epsilon wave. Correlational studies suggest surface ECG and epicardial mapping demonstrate ECG changes show abnormalities in intraventricular myocardium (parietal block) instead of a specialized conduction abnormality (septal block). Prolonged right precordial QRS and epsilon waves are major diagnostic features. Local prolonged QRS in V1-V3 to ≥ 110 ms is a sensitive and specific marker for diagnosing ARVD, accounting for 12 leads associated with increased risk of sudden cardiac death [12]. Less common ECG changes include P-waves > 2.5 mV, low voltage QRS in peripheral leads and T-wave inversion in inferior lead [12].
The task force criteria for diagnostic AVD recommends imaging methods for examining global or regional myocardial dysfunction and structural abnormalities. Table 5 summarizes Task Force criteria for imaging methods alongside major and minor visualized morphological global or localized dysfunction and structural alterations. Echocardiography
Table 5: ARVD Diagnostic by Imaging Methods by Major and Minor Criteria
Imaging Method |
Major Criteria |
Minor Criteria |
2D Echo |
Regional RV akinesia/dyskinesia or aneurysm and PLAX RVOT ≥ 32 mm/PSAX RVOT ≥ 36 mm/fractional area change ≤ 33 % |
Regional RV akinesia/dyskinesia and PLAX RVOT ≥ 29 < 32 mm, PSAX RVOT ≥ 32 < 36 mm or fractional areas change > 33% ≤ 40% |
MRI |
Regional RV akinesia/dyskinesia or dyssynchronous RV contraction and ratio of RV end-diastolic volume to BSA ≥ 110mL/m2 (male) or ≥ 100 mL/m2 (female) or RVEF ≤ 40% |
RV akinesia/dyskinesia or dyssynchronous RV contraction and ratio RV end diastolic volume to BSA ≥ 100 to < 100 mL/m2 (male) or ≥ 90 to <100mL/m2 (female) or RVEF >40% to ≤ 45%. |
RV Angiography |
Regional RV akinesia/dyskinesia or aneurysm |
None |
Source: Marcus et al., 2010, p.1535 [67].
Echocardiography is a widely used non-invasive imaging technique often always representing the first line imaging technique for assessing patients suspected to have ARVD or for screening family members to identify those developing clinical manifestations of ARVD [12]. Echocardiography also presents a method for serial examination to evaluate disease progression during follow up of ARVD patients. Echo is also an important differential diagnosis method for ARVD because it easily rules out other underlying causes of RV disorders especially Ebstein anomaly and atrial septal defect. Besides evaluation of RV wall motion and morphological abnormalities, quantitative Echo evaluation of RV including assessment of end-diastolic cavity dimensions, RV wall thickness, volume and function improves diagnostic efficacy. Such typical echo features may avoid the need for contrast angiography or even magnetic resonance imaging. However, borderline or normal Echo findings would warrant further examinations using other imaging techniques [12]. Figure 6 shows 2D echo parasternal long axis view of the RV findings in ARVD detecting the presence of typical inferior sub-tricuspid bulging.
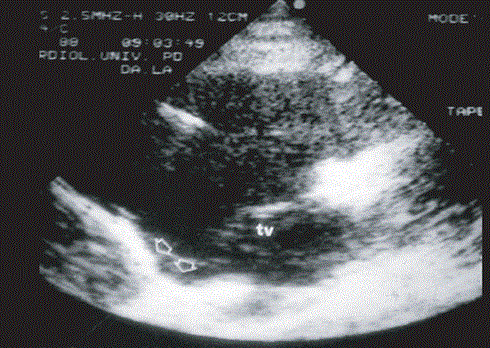
Figure 6: 2D Echo Findings of ARVD Patient
2-D echocardiography findings in ARVD. Presence of typical inferior sub-tricuspid bulging along parasternal long axis view of the RV: TV tricuspid valve. Source: Thiene et al., 2007, p.8 [1]
Cardiac magnetic resonance imaging (MRI) is an important non-invasive imaging tool capable of characterizing tissue and distinguishing fat from muscle tissue. Fast spin or turbo spin echo are recommended for black blood imaging, which best assesses the RV free wall and outflow tract. Stack of axial images should image the entire RV by using 6-8 slices at 1 cm intervals [79]. Table 6 summarizes the recommended cardiac MRI protocol for ARVD
Table 6: Recommended Protocol for Cardiac MRI for ARVD
Sequence |
Imaging Plane |
Parameters |
Comments |
Double inversion recovery turbo/fast spin echo axial (with/without fat suppression or short axis without fat suppression) |
Axial: 6-8 images centered on LV/RV
Short axis: 6-8 images centered on LV |
TR=2 R-R interval; TE=5ms; slice thickness = 5 mm; inter-slice gap = 5 mm FOV = 28-34 cm |
Optimal characterization of RV free wall
Fat suppression improves diagnosis of RV fat infiltration. |
Steady State Free Precession (SSFP) Bright Blood Cine Image |
Axial 4-chamber and short axis. Optional RV 3-chamber |
TR/TE minimum; flip angle = 450-700; slice thickness = 8 mm; inter-slice gap = 2 mm; Views/segment = 16-20 |
Axial images improves access to RV wall motion; RV quantitation on short axis cine images |
Contrast Gadolinium Administered Based on Institutional Protocol (0.15-0.2 mmol/kg) |
Inversion time (TI) Scout |
4-chamber |
|
Suppresses normal myocardium for right TI |
Late Gadolinium Enhanced (LGE) imaging |
Axial, short axis, 4-chmaber and vertical long axis |
TR/TE manufacturer recommendation; flip angle = 200-250; slice thickness = 8 mm; inter-slice gap = 2 mm; FOV =36-40 cm; use PSIR if available |
Best for imaging fibrosis and LV epicardia in inferolateral wall |
TI: Inversion Time; TE: Echo Time; TR: Repetition Time; FOV: Field of View; RV: Right Ventricle; LV: Let Ventricle
Source: Te Riele et al. 2014, p.4 [79]
The main challenge of cardiac MRI in imaging ARVD patients is the lack of well-defined sensitivity and specificity such that the quality of images is operator dependent and findings subject to individual interpretation (inter-observer variability) [20,79]. Marcus et al. [68] analyzed findings of American Multidisciplinary study on clinical presentation and diagnosis of ARVD and reports variation between MRI diagnosis of referral center and core laboratory. Core laboratory did not confirm 29% of diagnosis confirmed by the referral center [68]. Cine-MRI promises better detection by the ability to estimate RV volume and wall motion abnormalities due to akinesia, dyskinesia and aneurysms [1]. Figure 7 shows cardiac MRI four-chamber and short-axis imaging planes of ARVD patient with predominant RV abnormalities. The left panels show end-diastolic and right panel end-systolic. The arrow in the 4-chamber end-diastolic image shows subtricuspid dyskinesia while arrows in the short axis images show RV free wall aneurysm – systolic and diastolic bulging.
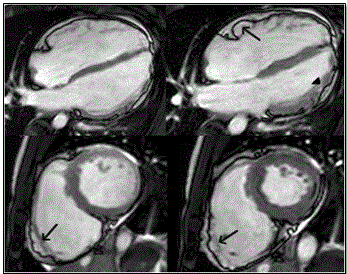
Figure 7: MRI - 4 Chamber (Top Panels) and Short Axis (Bottom Panels) or ARVD Heart
Top panel shows 4-chamberand bottom panels short-axis view of bright blood images in ARVD patient with predominant RV abnormalities. Left panels end-diastolic images and right panel end-systolic images. Sub-tricuspid dyskinesia in the end-systolic 4-chamber (arrow) and RV free wall aneurysms in both short axis images (arrows). Source: Te Riele et al. 2014, p.5 [79]
Further, Figure 8 shows MRI images of ARVD patient with predominant LV abnormalities. Bright blood images/LGE MRI (bottom panels) show dilated LV and mid-myocardial pattern in basal septum and basal lateral walls (arrows)
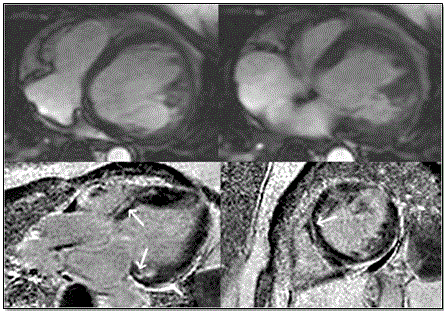
Figure 8: Top Panels – Horizontal Long Axis; Bottom Panels Bright Blood/LGE
Top panels show horizontal long axis bright blood and bottom panels show LGE images of ARVD patient having predominant LV abnormalities. There is dilated LV in the bright blood images. LGE is shown in a mid-myocardial pattern in basal septum and basal lateral walls (bottom panels arrows). Source: Te Riele et al. 2014, p.6 [79]
Many experts still consider right ventricular (RV) angiography as a diagnostic method of choice for assessing RV function [29]. The imaging technique provides angiographic evidence of akinesia or dyskinesia bulging in the triangle of dysplasia (Figure 1): infundibular, apical, and sub-tricuspid regions, with a high diagnostic specificity > 90% [14,69]. Prominent dilation akinesia accompanied by irregular aspect usually involving inferior RV wall have a significant association with ARVD diagnosis. Recommended RV angiography best evaluates by bi-plane angiography obtaining views in the 450 right anterior oblique and 450 left anterior oblique during sinus rhythm [29]. However, same to cardiac MRI, RV angiography suffers from significant inter-observer variability [12].
This systematic review and meta-analysis included studies that reported outcomes of various diagnosis methods for ARVD. Studies that did not include outcomes of diagnosis were excluded as well as studies with only abstracts that provided limited information on diagnostic outcomes. Online databases PubMed, EMBASE, Ovid MEDLINE, and Google Scholar were searched using the search strategy: “arrhythmogenic right ventricular dysplasia (ARVD)” OR “arrhythmogenic right ventricular cardiomyopathy (ARVC)” AND “diagnosis”. There was no time restriction or age of the patients used. All titles and abstracts of the studies were evaluated. After exclusion of studies based on titles and abstracts, full articles were evaluated and those meeting the inclusion criteria included. Additional studies were identified and reviewed using the same inclusion and exclusion criteria from screening bibliographies of the included studies. All the included studies were reviewed and relevant patient details and diagnostic features: method, measurement and findings were recorded in Microsoft Excel Worksheet. For, studies with duplicated data, details of the most recent study was recorded. Table 7 provides a summary of data from the included studies. Search Results
Table 7: Summary of Studies on Diagnosis of ARVD
1st Author [Ref. No.] |
Year |
Sample |
Method |
Measurement |
Findings |
Nasir et al. [83] |
2004 |
50 |
ECG |
Prolonged S-wave upstroke in V1-V3; Epsilon wave; prolonged QRS duration; T-wave inversion in V1-V3 |
A prolonged S-wave upstroke in V1 through V3 is a frequent ECG finding (95%) and diagnostic marker for ARVD. |
Peters et al. [84] |
2003 |
265 |
ECG |
Localized right precordial QRS prolongation (98%); Epsilon waves; T wave inversions |
ECG abnormalities are frequently encountered in ARVD patients. |
Peters et al. [85] |
2007 |
343 |
ECG |
Depolarization/ repolarization abnormalities |
Findings meet those of ECG diagnostic criteria by Task Force (WHO/ISFC/ESC) |
Peters et al. [86] |
2008 |
360 |
ECG |
QRS fragmentation Epsilon potential |
QRS fragmentation has the same diagnostic value as epsilon waves |
Cox et al. [87] |
2008 |
42 |
ECG |
Activation delay and VT |
Additional criteria improved the Task Force criteria on diagnosis of ARVD. |
Nava et L.[88] |
2000 |
138 |
SAECG |
SAECG abnormality (94.4%) |
A close correlation between SAECG abnormality and extent of disease |
Marcus et al. [6] |
1982 |
24 |
Echo/RV Angiography |
RV diastolic dimensions/ RV angiography |
Echo and RV angiography findings may provide composite clinical profile of ARVD patients |
Yoerger et al. [89] |
2005 |
29 |
Echo |
RV dimensions/ Fractional area change |
Echo can assess right atrial, RV chamber dimensions, RV regional function |
Robertson et al. [90] |
1985 |
10 |
Echo/RV Angiography |
RV enlargement |
Echo findings of diffuse RV enlargement may avoid the need for angiography |
Bomma et al. [65] |
2004 |
89 |
MRI |
Intramyocardial fat or wall thinning |
Re-evaluation of patients diagnosed using MRI did not confirm ARVD suggesting high cases of misdiagnosis using MRI |
Tandri et al [91] |
2006 |
40 |
MRI |
Inter-observer variability |
MRI assessment of RV structure/function are reproducible for experienced observer but fat infiltration lacks specificity and has low reproducibility. |
Bluemke et al. [92] |
2003 |
45 |
MRI |
RV/LV enlargement, RV abnormal morphology, size and location of fibrofatty depositions |
RV morphology and enlargement are reliable MRI markers for ARVD but fatty deposits have the greatest interobserver variability |
Tandri et al. [93] |
2003 |
12 |
MRI |
RV structural alteration and dysfunction |
MRI findings meets Task Force criteria in detecting intramyocardial fat, RV hypertrophy, RV-end diastolic diameter and volume, and ejection fraction. |
Tandri et al [94] |
2005 |
30 |
Delayed Enhanced -MRI |
Myocardial fibrofatty changes |
DE-MRI is able to detect fibrofatty changes in myocardium in ARVD with excellent correlation with histopathology findings. |
Ricci et al. [95] |
1992 |
8 |
MRI |
Myocardial fibrofatty changes |
MRI is useful in integrating Echo findings for diagnosis of ARVD |
Bomma et al. [96] |
2007 |
31 |
Multidetector computed tomography (MDCT) |
17/31 met Task Force criteria for ARVD diagnosis |
Cardiac MDCT has the potential to evaluate qualitative and quantitative RV changes in ARVD patients but limited in ICD and motion artefacts |
Asimaki et al. [97] |
2009 |
11 |
Immuno-histochemical analysis |
Mutation in desmosomal protein plakoglobin |
Immunoreactive signal levels for plakoglobin is higher in ARVD patients compared to normal subjects promising as novel diagnostic approach |
Chimenti et al. [98] |
2004 |
30 |
EMB |
Biopsy on apex, RV inferior/anterior free wall and septal/apical regions |
Diagnosis based on fatty tissues (30%) no difference in arrhythmic patterns, structural/functional in Echo/angiography. EMD is a reliable method to detect myocarditis mimicking ARVD. |
|
|
1557 |
|
|
|
*ECG: Electrocardiography; WHO: World Health Organization; ISFC: International Society & Federation of Cardiologist; ESC: European Society of Cardiologists; SAECG: Signal Averaged ECG; VT: Ventricular Tachycardia; EMB: Endomyocardial Biopsy
The search strategy identified 321 articles. After a review, exclusion and cross-referencing, a total of 18 studies were included in the systematic review and meta-analysis (Table 7). The 18 studies [6,65,83-98] published between 1982 and 2009 investigated various methods used in diagnosing ARVD. The studies investigated electrocardiography [6,83-90], echocardiography [6,89,90], cardiac MRI [65,91-95], cardiac computed tomography [96] and endomyocardial biopsy [97,98]. Overall, the 18 studies included 1,557 patients diagnosed with ARVD.
The findings reveal diagnosis of ARVD depends on a combination of methods assessing functional and structural alterations in the myocardium. The most common method was electrocardiography abnormalities adopted by 50% (half) of the studies, followed by cardiac MRI by 33.3% echocardiography by 16.7%, endomyocardial biopsy by 11% and finally cardiac computed tomography by 5.5%. the ECG method measured conduction (depolarization or repolarization) abnormalities such as Prolonged S-wave upstroke in V1-V3; Epsilon wave; prolonged QRS duration; T-wave inversion in V1-V3 [83],localized right precordial QRS prolongation (98%); Epsilon waves; T wave inversions [84], QRS fragmentation Epsilon potential [84,86]. Echocardiography assessed both functional and morphological myocardial alterations: RV diastolic dimension [6], RV dimensions and fractional area changes [89] and RV enlargement [90]. Cardiac MRI assessed morphological changes: myocardial fibrofatty changes [94,95], intramyocardial wall thinning [65] and RV enlargement/size and location of fibrofatty depositions [92]. Endomyocardial biopsy assessed fatty tissues but was more accurate for differential diagnosis to detect and exclude myocarditis, which mimics ARVD.
In the present analysis, diagnosis of ARVD lacks a gold standard method for characterization and the detection of fibrofatty tissues in the myocardium, which is the morphological hallmark of ARVD. As a result, various methods, both non-imaging methods – electrocardiography (ECG) and endomyocardial biopsy – and imaging methods – echocardiography, cardiac MRI and cardiac CT – play a role in the diagnosis. Diagnosis assesses both functional (conduction abnormalities) and morphological changes in myocardial mass). The findings reveal that the main target of ECG diagnosis of ARVD is detecting depolarization/repolarization abnormalities, which have been demonstrated in up to 98% of ARVD patients [84]. Further, signal-average ECG abnormality correlate with disease severity [88]. Echocardiography and angiography have been shown to detect changes in RV morphology and function. Echocardiography detection of diffuse RV enlargement could avoid the need for endomyocardial biopsy [90]. Endomyocardial biopsy on the other hand is not a very common method for diagnosing ARVD but it is reliable in differential diagnosis to discriminate myocarditis, which mimics ARVD [98]. Cardiac MRI is a reliable imaging modality in detecting intramyocardial fat, RV hypertrophy, RV-end diastolic diameter and volume, and ejection fraction as per the Task Force guidelines. However, cardiac MRI suffers from significant inter-observer variability especially in the detection of intramyocadial fat or wall thinning for ARVD diagnosis [65,91,92].
The use of multiple methods for ARVD diagnosis is consistent with the ESC/ISFC Task Force revised criteria for ARVD diagnosis [29]. The criteria emphasizes on the assessment of both global or regional dysfunction and morphological alteration using an appropriate imaging modality or a combination of imaging modalities, and repolarization and conduction abnormalities measured using electrocardiography. In addition to these methods, the assessment of arrhythmias and family history is essential to provide additional supportive diagnostic clues [9,66]. Despite the availability of the standardized criteria, concerns are emerging on the lack of defined quantifiable measures for myocardial dysfunction for echocardiography and cardiac MRI imaging modalities [68]. There is also the lack of large-scale studies examining and validating the diagnostic value of the ECS/ISFC Task Force criteria in the diagnosis of ARVD [67]. Moreover, although ARVD has been demonstrated to have a genetic basis, standard guidelines on the use of familial screening has not been established, which would have improved prediction of those at a higher risk of developing ARVD as well as improve the diagnosis of ARVD. In summary, although ARVD lacks a gold standard diagnostic method, a combination of ECG abnormalities, and characterization of functional and morphological features using echocardiography and cardiac MRI imaging g modalities have been recommended for ARVD diagnosis. Familial history and endomyocardial biopsy provide supportive diagnostic and differential diagnostic clues.
Whereas the ESC/IFSC Task Force has developed standardized diagnostic guidelines, ARVD lacks well-defined and widely accepted guidelines for clinical management. The current guidelines developed from a review of literature categorizes clinical management into symptomatic and asymptomatic patients, and with or without family history of ARVD. Figure 9 provides a schematic illustration of the proposed clinical management guidelines for ARVD.
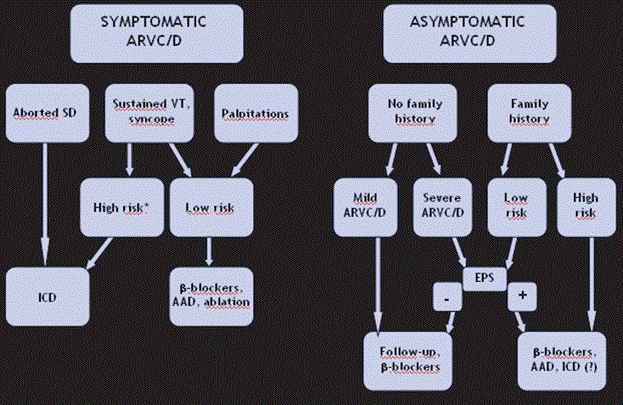
Figure 9: Proposed Algorithm for Clinical Management of ARVD
The proposed algorithm for clinical management of ARVD relies on the presence of symptomatic and asymptomatic ARVD. ARVD with aborted sudden cardiac death requires ICD while symptoms such as sustained VT, syncope and palpitations may require medical therapy or ablation. Asymptomatic patients with or without family history but with mild/severe ARVD may require medical therapy and follow-up. Source: Thiene et al., 2007, p.13 [1].
An important goal of clinical management of ARVD is to prevent sudden cardiac death. According to Thiene et al. [1], prevention of cardiac death in ARVD patients should focus on eradication of the disease using ICD, a reduction of sporting (exertional physical exercises) and the use of medical therapy to manage sustained arrhythmias. Figure 10 illustrates the four main clinical approaches to prevent sudden cardiac death in ARVD patients.
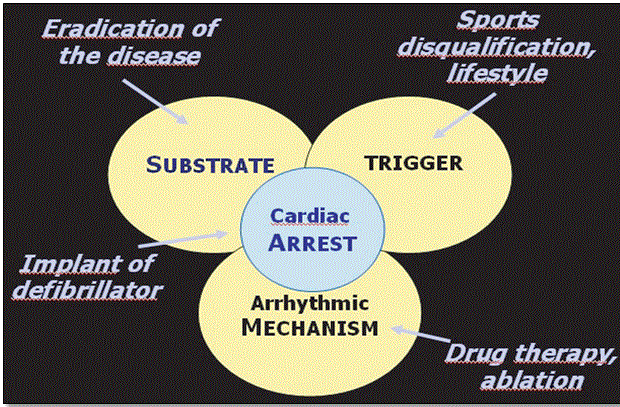
Figure 10: Levels for Prevention of Sudden Death in ARVD Patients
Prevention of sudden cardiac death in ARVD patients should focus on the three possible causes: substrate (eradicate disease or use ICD; trigger (stop physically exerting exercises) and arrhythmic mechanisms (use medical, therapy or ablation). Source: Thiene et al., 2007, p.13 [1].
Based on the proposed algorithm for clinical management of ARVD [1], the main approaches are genetic counselling, medical therapy, radiotherapy ablation and device therapy (ICD) as discussed hereinafter.
Genetic counselling is considered for patients diagnosed or suspected to have familial (inherited) ARVD. A genetic counsellor should be trained in medical genetics and counselling to assist physicians and patients in the genetic testing process. Counselling usually involves a review of diagnosis, detailed family history and cost, and discussion of tests carried out. Genetic testing assists patients to understand the underlying reasons causing the disease, prognosis and any risk the condition poses to close family members [34]. Genetic testing informs the consideration of prevention of triggers such as intense exercise activities [1].
Clinical findings predicting long-term outcome of ARVD patients remains unclear and exact guidelines to select patients requiring pharmacotherapy are lacking. Methods to predict prognosis of pharmacotherapy remains undefined or efficacy in the prevention of ARVD-associated sudden cardiac death unproven [20]. However, the few studies that have evaluated therapy for asymptomatic ARVD patients having focused on mild morphologic RV abnormalities in the absence of any discernible evidence of ventricular arrhythmias [29]. In these patients, β-adrenergic blocking agents are recommended for the reduction of possible adrenergic stimulated arrhythmias [29]. Pharmacotherapy using β-blockers and class I and III antiarrhythmic drugs is considered the initial treatment for ARVD patients with tolerated and mild ventricular arrhythmias [12,20]. In ARVD patients presenting with arrhythmias including non-sustained ventricular tachycardia, ventricular tachycardia or premature ventricular complex, or symptoms, the initial therapy should be β-blockers or antiarrhythmic drug therapy based on programmed ventricular stimulation with serial drug testing [20,29]. Current evidence indicates Sotalol or amiodarone (has both antiarrhythmic and β-blocking properties) alone or in combination with β-blockers show promising results with low proarrhythmic risk [80]. Although current efficacy of pharmacotherapy depends on symptom improvement, a more reliable approach should include treatment guidance by serial monitoring by 24-hour Holter monitoring and/or stress testing to demonstrate a reduction of arrhythmic events [29].
Radiofrequency ablation is indicated for patients unresponsive or intolerant to pharmacotherapy (especially antiarrhythmic drugs). However, radiofrequency ablation has shown frequent unsuccessfulness requiring multiple attempts [29]. Fontaine et al. [81] reports 32%, 45% and 66% success rates after the first, second and third ablation procedures in 50 patients unsuccessful with pharmacotherapy followed for a mean of 64 months. The main limitation of radiofrequency ablation is the progressive, diffuse and patchy nature of ARVD producing multiple arrhythmogenic foci [29].
Implantable cardioverter de?brillator (ICD) is recommended for ARVD patients with a high risk of sudden cardiac death including those resuscitated from cardiac arrest and those with a history of syncope and refractory life-threatening arrhythmias following pharmacotherapy. However, ICD has risks since regions of RV myocardium in ARVD patients is thin and non-contractile and has risk of penetration during ICD leads placement with subsequent tamponade, or fibrofatty substitution of myocardium leading to device in adequate sensing of arrhythmias amidst improper ICD failure in sensing ICD function or failure [12,2029]. Further, multivariate analysis suggests extensive RV dysfunction as an independent predictor of ICD discharge. Wichter et al. [82] finds adverse events in 37 patients during perioperative and follow up, and freedom from adverse events in 90%, 7%, 8% and 56% in 1, 3 and 5 years post implantation [82].
Clinical management of ARVD lacks standardized guidelines, which reduces efficacy of available treatment options. Although genetic counselling, medical therapy, ablation and device therapy have been recommended, their chronic effect on management of ARVD remains poorly understood. The present meta-analysis aims to review current clinical management methods to improve treatment efficacy and survival rates of ARVD patients.
Studies examining clinical management of ARVD and recorded parameters related to treatment outcomes of ARVD were searched and examined [1]. There was no time limitation or age of patients included in this meta-analysis. Electronic search engines used to identify relevant studies were PubMed, Scopus, Web of Science and Cochrane. Relevant medical subject headings (MESH) terms and a combination of key words used were arrhythmogenic right ventricular dysplasia (ARVD) or arrhythmogenic right ventricular cardiomyopathy (ARVC) AND “medication” OR “counselling” OR “ablation” OR “implantable Cardioverter Defibrillator [ICD]”.
Study selection used a three-level hierarchical search. The first level of selection included screening of authors, titles and abstracts. This first level selection excluded case studies (involving one or a few ARVD patients) animal studies and conference papers (still subject to revision) as well as compared abstracts to the inclusion criteria: (a) studies examined ARVD patients; (b) included at least one clinical management methods; and (c) reported outcomes on clinical management. Age of patients or time of publication was not a criteria for inclusion. After selection, relevant data was extracted from all the included studies. The extracted data was recorded in Microsoft Excel Worksheet. The extracted data included first author, year of publication, number of patients (sample), clinical end-pint studies, outcomes (survival/improvement) and a summary of study findings. Table 8 lists a summary of data on clinical management of ARVD from the included studies Study Characteristics
Table 8: Summary of Studies on ARVD Clinical Management
1st Author [Ref. No.] |
Year |
Sample |
Method |
Clinical End-point |
Survival/ Improvement |
Findings |
Wichter et al. [82] |
2004 |
60 |
ICD |
Event free survival |
49%, 30%, 26%, and 26% after 1, 3, 5, and 7 years |
Significant favorable long-term prognosis by ICD therapy for ARVD patients at risk of lethal arrhythmia. |
Roguin [99] |
2004 |
42 |
ICD |
Lead repositioning & systemic infection |
78% received appropriated ICD interventions |
ICD is well tolerated and important in managing ARVD |
Corrado et al. [100] |
2003 |
132 |
ICD |
Sudden death |
96% survival rate and 72% survival free RV fibrillation/flutter at 3 years. |
ICD provides life-saving protection by preventing lethal arrhythmias |
Link et al. [101] |
1997 |
12 |
ICD |
ICD perforation/sensing and efficacy |
33.3% had appropriate ICD therapy and one death after 1 month |
ICD with nonthoracotomy leads is safe and efficacious in ARVD patients. |
Bhonsale et al. [102] |
2011 |
84 |
ICD |
Survival free VT |
48% had appropriate ICD therapy in 4.7 years, 19% receiving intervention for RV fibrillation/flutter |
ICD provides life-saving protection in about half of ARVD patients. Non-sustained VT indicates appropriate ICD therapy |
Corrado et al. [103] |
2010 |
106 |
ICD |
Survival free |
At 58 months, 24% had appropriate ICD intervention and 16% with lethal VF/flutter |
ICD is an appropriate therapy for ARVD patients with no prior non-sustained VT or VF |
Komura et al. [104] |
2010 |
35 |
ICD /RFA |
Mortality/ hospitalization/VT recurrence |
ICD has lower VT recurrence and hospitalization but comparable morality rates to RFA |
ICD prevents hospitalization of ARVD patients with CHF onset by preventing VT. ICD with medication offers better treatment for ARVD patients |
Dalal et al. [105] |
2007 |
24 |
RFA |
Survival free VT |
VT survival free was 75%, 50%, and 25% after 1.5, 5, and 14 months |
There is a high recurrence rate of VT for patients undergoing RFA due to diffuse nature of ARVD and evolving electrical substrate |
Verma et al. [106] |
2005 |
22 |
RFA |
Survival free VT |
Short-term success in 82%. VT recurrence: 23%, 27%, and 47% after 1, 2, and 3 years |
RFA prevents VT in the short-term but recurrence increased over time |
Marcus et al. [107] |
2009 |
95 |
PT |
VT |
β-blockers have no significant effect on VT; Sotalol was associated risk of VT; Amiodarone had low risk of VT |
β-blockers/Sotalol offers no protection against VT while amiodarone has superior effect in preventing VT |
Lemery et al. [108] |
1989 |
12 |
PT |
VT |
At 7.9 years 11 patients (treated with antiarrhythmic drugs alone or with β-blockers) are alive |
Despite recurrent VT, ARVD patients have favorable prognosis when treated with pharmacotherapy |
Tonet et al. [109] |
1988 |
20 |
PT |
VT |
β-blockers/ amiodarone/ antiarrhythmic drugs with PES manages VT |
β-blockers and chronic amiodarone therapy have synergistic effect and together with PES inhibit VT |
ICD: Implantable Cardioverter De?brillator; VT: Ventricular Tachycardia; RFA: Radiofrequency Ablation; VF: Ventricular Fibrillation; PT: Pharmacotherapy; PES: Programmed Electrical Stimulation
After a review of titles, abstract and reading of the entire article against the inclusion/exclusion criteria, a total of 12 studies investigating various clinical management methods of ARVD patients were included for analysis. The studies were published between 1988 and 2011. The bulk of the studies (50%) examined implantable cardioverter de?brillator (ICD) [82,99-104] while the remaining examined radiofrequency ablation (25%) [104-106], and pharmacotherapy (25%) [107-109] In total, the 12 studies investigated 644 ARVD patients.
The seven studies on ICD therapy demonstrate ARVD patients have good toleration and the therapy provides long-term life-saving protection by preventing lethal ventricular arrhythmias including ventricular tachycardia and ventricular fibrillation or flutter. Further, ICD with non-thoracotomy leads is safe and efficacious in ARVD patients by reducing cases of perforation [101]. Whereas radiofrequency ablation has favorable short-term outcomes by preventing ventricular tachycardia (VT), there is significance recurrence of VT in the long-term because of diffuse nature of ARVD and evolving electrical substrate [104-106]. Finally, pharmacotherapy involving β-blockers and antiarrhythmic drugs offer favorable prognosis in the short-term [107-109]. Amiodarone has been shown to have superior protective effect against VT compared to β-blockers and Sotalol [107]. β-blockers taken with amiodarone and programmed electrical stimulation offer better protection against VT [109]. Finally, ICD together with pharmacotherapy provides long-term life-saving protection against VT [104].
Although ARVD lacks well-established guidelines for clinical management, proposed guidelines recommend management based on the presence or absence of symptoms. The present findings confirm that current research and clinical practice emphasize on treatment of symptoms. The main clinical management methods are device therapy (ICD), pharmacotherapy, and radiofrequency ablation. The main debilitating symptom of ARVD is lethal ventricular arrhythmias (tachycardia, fibrillation or flutter), which is the focus of current therapies. Consistent with findings of Thiene and associates [1], lethal arrhythmias are the principal cause of sudden cardiac death and the main therapeutic target for ARVD patients. ICD, medical therapy and RF ablation all focus on managing lethal arrhythmias. However, while RF ablation and medical therapy using beta-blockers and antiarrhythmic drugs are effective in the short-term, their efficacy diminishes with time, reducing their clinical value when chronic treatment is considered. RF ablation although effective in the short-term, suffers serious limitation of the multiple attempts with success rates achieved in the subsequent procedures [29,81]. On the other hand, ICD has been shown to be effective in both short term and long-term but given concomitant with medication, ICD promises chronic life-time protection against lethal ventricular arrhythmias and survival.
Whereas lethal arrhythmias are the main therapeutic target for clinical management, considering other factors such as etiology, physical exertion and genetic predisposition is also essential to improve clinical management. According to the proposed clinical management guidelines, aborted sudden cardiac death, palpitations, and family history are important additional features to inform the most appropriate treatment [1]. Marcus et al. [34] report of the importance of family screening or genetic counselling to unearth family history, which is clinically useful to provide additional clues to confirm the underlying cause and identify immediate family members at an increased risk of developing ARVD. Genetic testing also provides the need for additional tests and the reason for stopping physically exerting exercises that are a major trigger for sudden cardiac death. In summary, clinical management of ARVD focuses on treatment and management of arrhythmias as well as the prevention of triggers such as halting physically exerting exercises. However, the value of stopping physical exercises has not been demonstrated to convey protective effect against sudden cardia death or the development of arrhythmias.
Arrhythmogenic right ventricular dysplasia (ARVD) is a myocardial disorder characterized by pathological substitution of RV free wall by fibrofatty tissues. It is a rare condition affecting approximately 1 tin every 2,000 to 5,000 people. The clinical course of the disease includes four main phases: asymptomatic subclinical phase, followed by a symptomatic overt electrical disorder phase leading to RV failure phase and eventually bi ventricular failure phase. The main reasons for admission are sustained or recurrent lethal ventricular arrhythmias, symptomatic heart failure and sudden cardiac death. The predominant risk factors predisposing to the development of ARVD are young age, physically exerting exercise or sporting activities, malignant family history, extensive right ventricular disease with reduced LV ejection fraction, and LV ventricular involvement, syncope and ventricular tachycardia. ARVD has a predominant genetic etiology mainly mutations in desmosomal genes but also environmental factors may cause the disorder or facilitate genetic expression. The main predictors of poor prognosis are life-threatening arrhythmias, aborted sudden cardiac death or heart failure. Diagnosis uses multiple methods including endomyocardial biopsy, electrocardiography, echocardiography, RV angiography, cardiac MRI as recommended by Task Force criteria for diagnosis of ARVD. The disorder has no single therapy but clinical management focuses on preventing life-threatening arrhythmias and sudden cardiac death. Pharmacotherapy and radiofrequency ablation have favorable short-term outcomes while ICD (more so given concomitant with medical therapy) has long-term survival free from ventricular tachycardia. To improve clinical management of ARVD, there is need to define guidelines for predicting prognosis and long-term efficacy of current therapies.
References
- Thiene G, Corrado D, Basso C (2007) Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Orphanet Journal of Rare Diseases 2: 45.
- Dalla VS, Battaglia G, Zerbini E (1961) Auricularization' of right ventricular pressure curve. Am Heart J 61: 25-33. [Crossref]
- Fontaine GH (1977) Stimulation studies and epicardial mapping in ventricular tachycardia; study of mechanisms and selection for surgery. Reentrant Arrhythmias 334.
- Marcus FI, Fontaine G (1995) Arrhythmogenic right ventricular dysplasia cardiomyopathy: a review. Pacing and Clinical Electrophysiology 18: 1298-1314.
- Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, et al. (1997) Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. Journal of the American College of Cardiology 30: 1512-1520.
- Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, et al. (1982) Right ventricular dysplasia: a report of 24 adult cases. Circulation 65: 384-398. [Crossref]
- Thiene G, Nava A, Corrado D, Rossi L, Pennelli N (1988) Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 318: 129-133. [Crossref]
- Basso C, Thiene G, Corrado D, Angelini A, Nava A et al. (1996) Arrhythmogenic right ventricular cardiomyopathy. Circulation 94: 983-991.
- Richardson P, McKenna W, Bristow M, et al. (1996) Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation 93, 841-842
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, et al. (2007) Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. European Heart Journal 29: 270-276.
- Anderson EL (2006) Arrhythmogenic right ventricular dysplasia. American Family Physician 73: 1391-1398.
- Corrado D, Basso C, Thiene G (2000) Arrhythmogenic right ventricular cardiomyopathy: diagnosis, prognosis, and treatment. Heart 83: 588-595.
- Norman MW, McKenna WJ (1999) Arrhythmogenic right ventricular cardiomyopathy: perspectives on disease. German Journal of Cardiology 88: 550-554.
- Nava A, Thiene G, Canciani B, Scognamiglio R, Daliento, L, et al. (1988). Familial occurrence of right ventricular dysplasia: a study involving nine families. Journal of the American College of Cardiology, 12(5), 1222-1228.
- Peters, S, Trümmel M, Meyners W (2004) Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. International journal of cardiology 97: 499-501.
- Nava A, Bauce B, Basso C, Muriago M, Rampazzo A, et al. (2000) Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. Journal of the American College of Cardiology 36: 2226-2233.
- Corrado D, Basso C, Pavei A, Michieli P, Schiavon M, et al. (2006) Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. Jama 296: 1593-1601.
- Basso C, Corrado D, Marcus FI, Nava A, Thiene G (2009) Arrhythmogenic right ventricular cardiomyopathy. Lancet 373: 1289-1300. [Crossref]
- Thiene G, Nava A, Angelini A, Daliento L, Scognamiglio R, et al. (1990) Anatomoclinical aspects of arrhythmogenic right ventricular cardiomyopathy. Advances in Cardiomyopathies 397-408.
- Corrado D, Fontaine G, Marcus FI, McKenna WJ, Nava A, et al. (2000) Arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 101: e101-e106.
- Marcus FI, Fontaine GH, Frank R, Gallagher JJ, Reiter MJ (1989) Long-term follow-up in patients with arrhythmogenic right ventricular disease. European Heart Journal 10: 68-73.
- Pinamonti B, Di Lenarda A, Sinagra G, Silvestri F, Bussani R, et al. (1995) Long-term evolution of right ventricular dysplasia-cardiomyopathy. American heart journal 129: 412-415.
- Kullo IJ, Edwards WD, Seward JB (1995) Right ventricular dysplasia: the Mayo Clinic experience. Mayo Clin Proc 70: 541-548. [Crossref]
- Calkins H (2006) Arrhythmogenic right-ventricular dysplasia/cardiomyopathy. Current Opinion in Cardiology 21: 55-63.
- Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, et al. (2005) Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation 112: 3823-3832. [Crossref]
- Nasir K, Bomma C, Tandri H, Roguin A, Dalal D, et al. (2004) Electrocardiographic features of arrhythmogenic right ventricular dysplasia/cardiomyopathy according to disease severity. Circulation 110: 1527-1534.
- Calkins H (2008) Arrhythmogenic right ventricular dysplasia. Transactions of the American Clinical and Climatological Association,119: 273.
- Jaoude SA, Leclercq JF, Coumel P (1996) Progressive ECG changes in arrhythmogenic right ventricular disease Evidence foran evolving disease. European Heart Journal 17: 1717-1722.
- Gemayel C, Pelliccia A,Thompson PD (2001) Arrhythmogenic right ventricular cardiomyopathy. Journal of the American College of Cardiology 38: 1773-1781.
- Peters S, Peters H, Thierfelder L (1999) Risk stratification of sudden cardiac death and malignant ventricular arrhythmias in right ventricular dysplasia-cardiomyopathy. International Journal of Cardiology 71: 243-250.
- Maron BJ, Shirani J, Poliac LC, Mathenge R, Roberts WC, et al. (1996) Sudden death in young competitive athletes: clinical, demographic, and pathological profiles. Jama, 276: 199-204.
- Thiene G, Basso C, Corrado D (1999) Is prevention of sudden death in young athletes feasible? Cardiologia 44: 497-505. [Crossref]
- Pinamonti B, Miani D, Sinagra G, Bussani R, Silvestri, F, et al. (1996) Familial right ventricular dysplasia with biventricular involvement and inflammatory infiltration. Heart Muscle Disease Study Group. Heart 76: 66-69.
- Marcus FI, Edson S, Towbin JA (2013) Genetics of arrhythmogenic right ventricular cardiomyopathy. Journal of the American College of Cardiology 61: 1945-1948.
- James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, et al. (2013) Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/Cardiomyopathy–Associated desmosomal mutation carriers. Journal of the American College of Cardiology 62: 1290-1297.
- Hermida JS, Minassian A, Jarry G, Delonca J, Rey JL, et al. (1997) Familial incidence of late ventricular potentials and electrocardiographic abnormalities in arrhythmogenic right ventricular dysplasia. The American journal of cardiology 79: 1375-1380.
- Marcus F, Towbin JA, Zareba W, Moss A, Calkins H, Brown M, Gear K (2003) Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C). Circulation 107: 2975-2978.
- Awad MM, Calkins H, Judge DP (2008) Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nature clinical practice. Cardiovascular Medicine 5: 258.
- Thiene G, Corrado D, Nava A, Rossi L, Poletti A, Boffa GM, et al. (1991) Right ventricular cardiomyopathy: is there evidence of an inflammatory aetiology? European heart journal, 12: 22-25.
- Basso C, Thiene G (2005) Adipositas cordis, fatty infiltration of the right ventricle, and arrhythmogenic right ventricular cardiomyopathy. Just a matter of fat? Cardiovascular Pathology 14: 37-41.
- Burke AP, Farb A, Tashko G, Virmani R (1998) Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium. Circulation 97: 1571-1580.
- Sen Chowdhry S, Morgan RD, Chambers JC, McKenna WJ (2010) Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annual Review of Medicine 61: 233-253.
- Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, et al. (2008) Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. The American Journal of Human Genetics 82: 809-821.
- Ohno S (2016) The genetic background of arrhythmogenic right ventricular cardiomyopathy. Journal of Arrhythmia 32: 398-403.
- Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, et al. (2002) Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. The American Journal of Human Genetics 71: 1200-1206.
- Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, et al. (2007) A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. The American Journal of Human Genetics 81: 964-973.
- Lahtinen AM, Lehtonen A, Kaartinen M, Toivonen L, Swan H, et al. (2008) Plakophilin-2 missense mutations in arrhythmogenic right ventricular cardiomyopathy. International Journal of Cardiology 126: 92-100.
- Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, et al. (2010) Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. Journal of the American College of Cardiology 55: 587-597.
- Kapplinger JD, Landstrom AP, Salisbury BA, Callis TE, Pollevick GD, et al. (2011) Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia–associated mutations from background genetic noise. Journal of the American College of Cardiology 57: 2317-2327.
- Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. (2001) Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103: 196-200.
- Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, et al. (2001) Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2) Human Molecular Genetics 10: 189-194.
- Beffagna,G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C (2005) Regulatory mutations in transforming growth factor-ß3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovascular Research 65: 366-373.
- Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, et al. (2006) Suppression of canonical Wnt/ß-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. Journal of Clinical Investigation 116: 2012.
- Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, et al. (2006) Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation research 99: 646-655.
- Corrado D, Basso C, Rizzoli G, Schiavon M. Thiene G (2003) Does sports activity enhance the risk of sudden death in adolescents and young adults?. Journal of the American College of Cardiology 42: 1959-1963.
- Corrado D, Basso C, Schiavon M, Thiene G (2006) Does sports activity enhance the risk of sudden cardiac death? Journal of Cardiovascular Medicine 7: 228-233.
- Ananthasubramaniam K, Khaja F (1998) Arrhythmogenic right ventricular dysplasia/cardiomyopathy: Review for the clinician. Progress in Cardiovascular Diseases 41: 237-246.
- Miani D, Pinamonti B, Bussani R, Silvestri F, Sinagra G (1993) Right ventricular dysplasia: a clinical and pathological study of two families with left ventricular involvement. Heart 69: 151-157.
- Fontaine G, Fontaliran F, Lascault G, Frank R, Tonet J, et al. (1990) Congenital and acquired right ventricular dysplasia. Arch Mal Coeur Vaiss 83: 915-920. [Crossref]
- Grumbach IM, Heim A, Vonhof S, Stille SM, Mall G, Gonska (1998) Coxsackievirus genome in myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Cardiology 89: 241-245.
- Williams GT, Smith CA (1993) Molecular regulation of apoptosis: genetic controls on cell death. Cell 74: 777-779. [Crossref]
- Mallat Z, Tedgui A, Fontaliran F, Frank R, Durigon M, et al. (1996) Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N Engl J Med 335: 1190-1196. [Crossref]
- D'Amati G, Di Gioia CR, Giordano C, Gallo P (2000) Myocyte trans differentiation: a possible pathogenetic mechanism for arrhythmogenic right ventricular cardiomyopathy. Archives of Pathology Laboratory Medicine 124: 287-290.
- Bomma C, Rutberg J, Tandri H, Nasir K, Roguin A, et al. (2004) Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Journal of Cardiovascular Electrophysiology 15: 300-306.
- McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom LC (1994) Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. British Heart Journal 71: 215.
- Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B (2010) Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 121: 1533-1541.
- Marcus FI, Zareba W, Calkins H, Towbin JA, Basso C, et al. (2009) Arrhythmogenic right ventricular cardiomyopathy/dysplasia clinical presentation and diagnostic evaluation: Results from the North American Multidisciplinary Study. Heart Rhythm 6: 984-992.
- Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, et al. (2011) HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 13: 1077-1109.
- Zhang M, Tavora F, Oliveira JB, Li L, Franco M (2012) PKP2 mutations in sudden death from arrhythmogenic right ventricular cardiomyopathy (ARVC) and sudden unexpected death with negative autopsy (SUDNA). Circulation Journal 76: 189-194.
- Zhang M, Xue A, Shen Y, Oliveira JB, Li L, et al. (2015) Mutations of desmoglein-2 in sudden death from arrhythmogenic right ventricular cardiomyopathy and sudden unexplained death. Forensic Science International 255: 85-88.
- Murray B (2012) Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C): a review of molecular and clinical literature. Journal of Genetic Counseling 21: 494-504.
- Lahtinen AM, Lehtonen E, Marjamaa A, Kaartinen M, Heliö T (2011) Population-prevalent desmosomal mutations predisposing to arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 8: 1214-1221.
- Mestroni L, Taylor MR (2011) Hearing the noise the challenges of human genome variation in genetic testing. J Am Coll Cardiol 57: 2328-2329. [Crossref]
- Den Haan AD, Tan BY, Zikusoka MN, Lladó LI, Jain R, et al. (2009) Comprehensive desmosome mutation analysis in North Americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circgenetics 4: 109.
- Christensen AH, Benn M, Bundgaard H, Tybjærg HA, Haunso S, et al. (2010) Wide spectrum of desmosomal mutations in Danish patients with arrhythmogenic right ventricular cardiomyopathy. Journal of Medical Genetics 47: 736-744.
- Barahona DC, Benito B, Campuzano O, Iglesias A, Leung TL, et al. (2010) Role of genetic testing in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Clinical Genetics, 77: 37-48.
- Bauce B, Nava A, Beffagna G, Basso C, Lorenzon A, et al. (2010) Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart rhythm 7: 22-29.
- Te Riele AS, Tandri H, Bluemke DA (2014) Arrhythmogenic right ventricular cardiomyopathy (ARVC): Cardiovascular magnetic resonance update. Journal of Cardiovascular Magnetic Resonance 16: 50.
- Wichter T, Hindricks G, Lerch H, Bartenstein P, Borggrefe M, et al. (1994) Regional myocardial sympathetic dysinnervation in arrhythmogenic right ventricular cardiomyopathy. An analysis using 123I-meta-iodobenzylguanidine scintigraphy. Circulation 89: 667-683.
- Fontaine G, Tonet J, Gallais Y, Lascault G, Hidden LF, et al. (2000) Ventricular tachycardia catheter ablation in arrhythmogenic right ventricular dysplasia: a 16-year experience. Current Cardiology Reports 2: 498-506.
- Wichter T, Paul M, Wollmann C, Acil T, Gerdes P, et al. (2004) Implantable cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy. Circulation 109: 1503-1508.
- Nasir K, Bomma C, Tandri H, Roguin A, Dalal D, et al. (2004) Electrocardiographic features of arrhythmogenic right ventricular dysplasia/cardiomyopathy according to disease severity. Circulation 110: 1527-1534.
- Peters S, Trümmel M (2003) Diagnosis of arrhythmogenic right ventricular dysplasia-cardiomyopathy: value of standard ECG revisited. Annals of Noninvasive Electrocardiology 8: 238-245.
- Peters S, Trümmel M, Koehler B, Westermann KU (2007) The value of different electrocardiographic depolarization criteria in the diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Journal of Electrocardiology 40: 34-37.
- Peters S, Trümmel, M, Koehler B (2008) QRS fragmentation in standard ECG as a diagnostic marker of arrhythmogenic right ventricular dysplasia–cardiomyopathy. Heart Rhythm 5: 1417-1421.
- Cox MG, Nelen MR, Wilde AA, Wiesfeld AC, SMAGT J (2008) Activation delay and VT parameters in arrhythmogenic right ventricular dysplasia/cardiomyopathy: toward improvement of diagnostic ECG criteria. Journal of Cardiovascular Electrophysiology 19: 775-781.
- Nav A, Folino AF, Bauce B, Turrini P, Buja GF, et al. (2000) Signal-averaged electrocardiogram in patients with arrhythmogenic right ventricular cardiomyopathy and ventricular arrhythmias. European heart journal 21: 58-65.
- Yoerger DM, Marcus F, Sherrill D, Calkins H, Towbin JA (2005) Multidisciplinary Study of Right Ventricular Dysplasia Investigators Echocardiographic findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia: new insights from the multidisciplinary study of right ventricular dysplasia. Journal of the American College of Cardiology 45: 860-865.
- Robertson JH, Bardy GH, German LD, Gallagher JJ, Kisslo J (1985) Comparison of two-dimensional echocardiographic and angiographic findings in arrhythmogenic right ventricular dysplasia. The American Journal of Cardiology 55: 1506-1508.
- Tandri H, Castillo E, Ferrari VA, Nasir K, Dalal D (2006) Magnetic resonance imaging of arrhythmogenic right ventricular dysplasia: sensitivity, specificity, and observer variability of fat detection versus functional analysis of the right ventricle. Journal of the American College of Cardiology, 48: 2277-2284.
- Bluemke DA, Krupinski EA, Ovitt T, Gear K, Unger E, et al. (2003) MR Imaging of arrhythmogenic right ventricular cardiomyopathy: morphologic findings and interobserver reliability. Cardiology 99: 153-162.
- Tandri H, Calkins H, Nasir K, Bomma C, Castillo E, et al. (2003) Magnetic resonance imaging findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia. Journal of cardiovascular electrophysiology 14: 476-482.
- Tandri H, Saranathan M, Rodriguez ER, Martinez C, Bomma C, et al. (2005) Noninvasive detection of myocardial fibrosis in arrhythmogenic right ventricular cardiomyopathy using delayed-enhancement magnetic resonance imaging. Journal of the American College of Cardiology 45: 98-103.
- Ricci C, Longo R, Pagnan L, Dalla PL, Pinamonti B, et al. (1992) Magnetic resonance imaging in right ventricular dysplasia. The American Journal of Cardiology 70: 1589-1595.
- Bomma C, Dalal D, Tandri H, Prakasa K, Nasir K, et al. (2007) Evolving role of multidetector computed tomography in evaluation of arrhythmogenic right ventricular dysplasia/cardiomyopathy. The American Journal of Cardiology 100: 99-105.
- Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, et al. (2009) A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. New England Journal of Medicine 360: 1075-1084.
- Chimenti C, Pieroni M, Maseri A, Frustaci A (2004) Histologic findings in patients with clinical and instrumental diagnosis of sporadic arrhythmogenic right ventricular dysplasia. Journal of the American College of Cardiology, 43: 2305-2313.
- Roguin A, Bomma CS, Nasir K, Tandri H, Tichnell C, et al. (2004) Implantable cardioverter-defibrillators in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Journal of the American College of Cardiology 43: 1843-1852.
- Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, et al. (2003). Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 108: 3084-3091.
- Link MS, Wang PJ, Haugh CJ, Homoud MK, Foote CB, et al. (1997). Arrhythmogenic right ventricular dysplasia: clinical results with implantable cardioverter defibrillators. Journal of Interventional Cardiac Electrophysiology 1: 41-48.
- Bhonsale A, James CA, Tichnell C, Murray B, Gagarin D (2011) Incidence and predictors of implantable cardioverter-defibrillator therapy in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy undergoing implantable cardioverter-defibrillator implantation for primary prevention. Journal of the American College of Cardiology 58: 1485-1496.
- Corrado D, Calkins H, Link MS, Leon L, Favale S, et al. (2010) Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation 122: 1144-1152.
- Komura M, Suzuki JI, Adachi S, Takahashi A, Otomo K, et al. (2010) Clinical course of arrhythmogenic right ventricular cardiomyopathy in the era of implantable cardioverter-defibrillators and radiofrequency catheter ablation. International heart journal 51: 34-40.
- Dalal D, Jain R, Tandri H, Dong J, Eid SM, et al. (2007) Long-term efficacy of catheter ablation of ventricular tachycardia in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Journal of the American College of Cardiology 50: 432-440.
- Verma A, Kilicaslan F, Schweikert RA, Tomassoni G, Rossillo A, et al. (2005). Short-and long-term success of substrate-based mapping and ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia. Circulation 111: 3209-3216.
- Marcus GM, Glidden DV, Polonsky B, Zareba W, Smith LM (2009) Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: a report from the North American ARVC Registry. Journal of the American College of Cardiology 54: 609-615.
- Lemery R, Brugada P, Janssen J, Cheriex E, Dugernier T, et al. (1989) Nonischemic sustained ventricular tachycardia: clinical outcome in 12 patients with arrhythmogenic right ventricular dysplasia. Journal of the American College of Cardiology 14: 96-105.
- Tonet J, Frank R, Fontaine G, Grosgogeat Y (1988) Efficacy and Safety of Low Doses of Beta-Blocker Agents Combined with Amiodarone in Refractory Ventricular Tachycardia. Pacing and Clinical Electrophysiology 11: 1984-1989.