Abstract
Yamaguchi syndrome, also known as apical hypertrophic cardiomyopathy (AHCM), is an uncommon kind of hypertrophic cardiomyopathy that is primarily observed in East Asia, with only limited data available from Latin America. We describe a 68-year-old man from Latin America who had hypertension, frequent palpitations, and prolonged ventricular tachycardia. Apical enlargement with widespread fibrosis and the traditional "ace of spades" appearance were verified by imaging. Electrophysiological mapping localized the arrhythmia to the mitral posteroseptal papillary muscle, and successful radiofrequency ablation was performed. An implantable cardioverter-defibrillator (ICD) was placed for primary prevention, despite negative genetic testing. This case underscores that AHCM is not geographically exclusive, may pose significant arrhythmic risk, and requires comprehensive evaluation using advanced imaging and multidisciplinary care.
Keywords
ventricular tachycardia, yamaguchi syndrome, apical hypertrophic cardiomyopathy, ablation, implantable cardioverter desfibrillator
Introduction
Apical hypertrophic cardiomyopathy (AHCM) is a very rare medical condition first described in 1976 by Sakamoto, et al. [1], and later in 1979, Yamaguchi described the echocardiographic and ventriculographic findings [2]. This type of cardiomyopathy is characterized by asymmetric apical hypertrophy in the absence of other secondary causes [1,3]. It is a primary and usually familial cardiac disorder with a very heterogeneous presentation. This apical phenotype remains very rare compared to the rest of hypertrophic cardiomyopathy (HCM); its prevalence is not known exactly. It is known that Japanese patients are more predisposed to develop it compared to patients in the USA (15% vs. 3%, respectively) [4,5]. There are very few studies in Latin America. Recent work in some regions of Colombia in patients with HCM found that the most frequent left ventricle hypertrophy phenotype was asymmetric (64.9%), followed by concentric (28.1%) and much less frequent apical (7.0%) [6]. It’s prevalence remains low, but it is already known that it is not a pathology that exclusively affects Asians.
Case presentation
A 68-year-old man with a history of systemic arterial hypertension, under treatment with bisoprolol, with no family history of premature coronary artery disease or sudden cardiac death. The patient consulted for frequent episodes of "palpitations" of long duration. During outpatient follow-up, a cardiovascular stress test was requested, presenting dyspnea, monofocal Premature ventricular contraction and sustained ventricular tachycardia (VT) at maximum effort (Figure 1A). Because VT is a risk marker for sudden death, the patient was hospitalized. During his stay in the emergency department, an electrocardiogram was performed and showed sinus rhythm with T-wave inversion from V2 to V6 (Figure 1B). Transthoracic echocardiography showed left ventricular hypertrophy of apical predominance (Figure 1C), with a polar Strain map showing abnormal longitudinal function in the apical segments (Figure 1D).
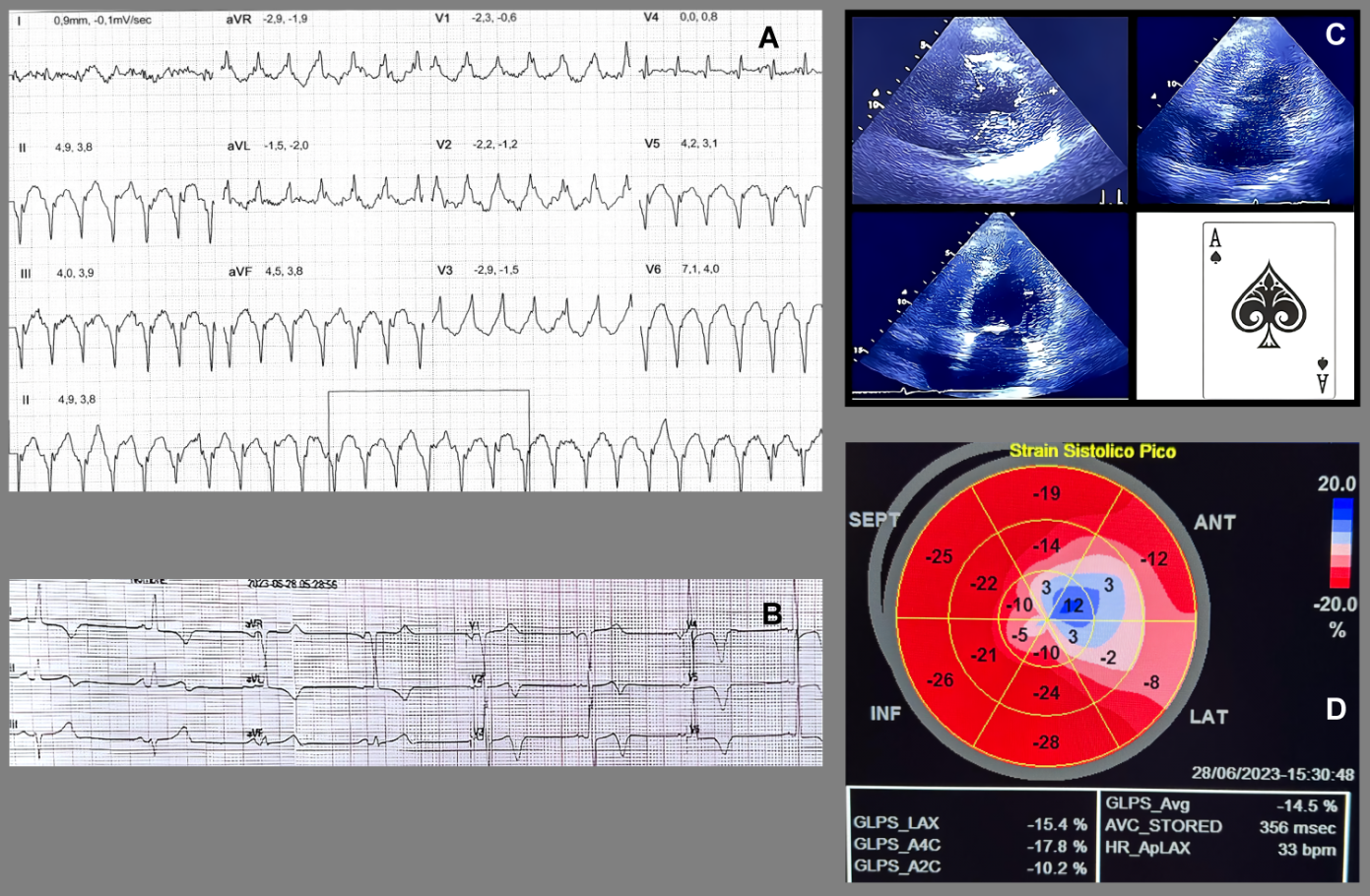
Figure 1. (A): Stress test with report of sustained ventricular tachycardia (VT) in maximal stress event with unified ventricular duplets. (B): Electrocardiogram showing sinus rhythm with marked wide-based negative T-wave inversion of V2-V6. (C): Transthoracic echocardiogram short axis view and four chambers with increased thickness at the apical level, also note the "Ace of Spades" shape in the apical segment and the decrease in the interventricular volume. (D): Polar map of global longitudinal myocardial deformation showing involvement of the apical segments with a tendency towards the anterolateral and medial inferolateral segment
Intramyocardial enhancement was observed in the apical third with an anterior, inferior lateral and septal patchy-diffuse pattern, all compatible with apical hypertrophic cardiomyopathy with preserved biventricular function and a non-ischemic myocardial enhancement pattern, suggestive of diffuse fibrosis in hypertrophic areas of the left ventricle, with a semiquantitative calculation of LV myocardial fibrosis of 52% (Figure 2A). The 3-chamber magnetic resonance (MRI) imaging slice showed a left ventricle (LV) with an end-diastolic volume (EDV) of 63 ml and an end-systole volume (ESV) of 23 ml, with a sign of "ace of spades" due to collapse in systole in the apical third of the ventricle with a maximum thickening at the apical level of 23 mm (Figure 2B). It was decided to perform a left heart catheterization due to electrocardiographic findings and episodes of ventricular tachycardia; this showed coronary arteries without lesions. Due to findings in the stress test of VT and high load of diffuse fibrosis in hypertrophic areas in the MRI, it was decided to take the patient to an electrophysiological study to determine the origin of the arrhythmia and substrate arrhythmic modulation through three-dimensional electroanatomical mapping system. With identification of PVC and focal VT from the mitral postero-septal papillary muscle (Figure 3A), so radiofrequency ablation was performed successfully (Figure 3B). Subsequently, it was decided to place a definitive percutaneous bicameral cardiodesfibrillator to prevent sudden death. The patient tolerated the procedure without complications and was prescribed amiodarone 200 mg orally daily during 3 months. Studies for Fabry disease were requested which were negative, and a panel for MYH7, TNNI3, GLA, MYBPC3, TNNT2, TPM1, MYL2, MYL3, LAMP3, LSAMP, PRKAG2, АСТАА1 gene mutations with negative result.
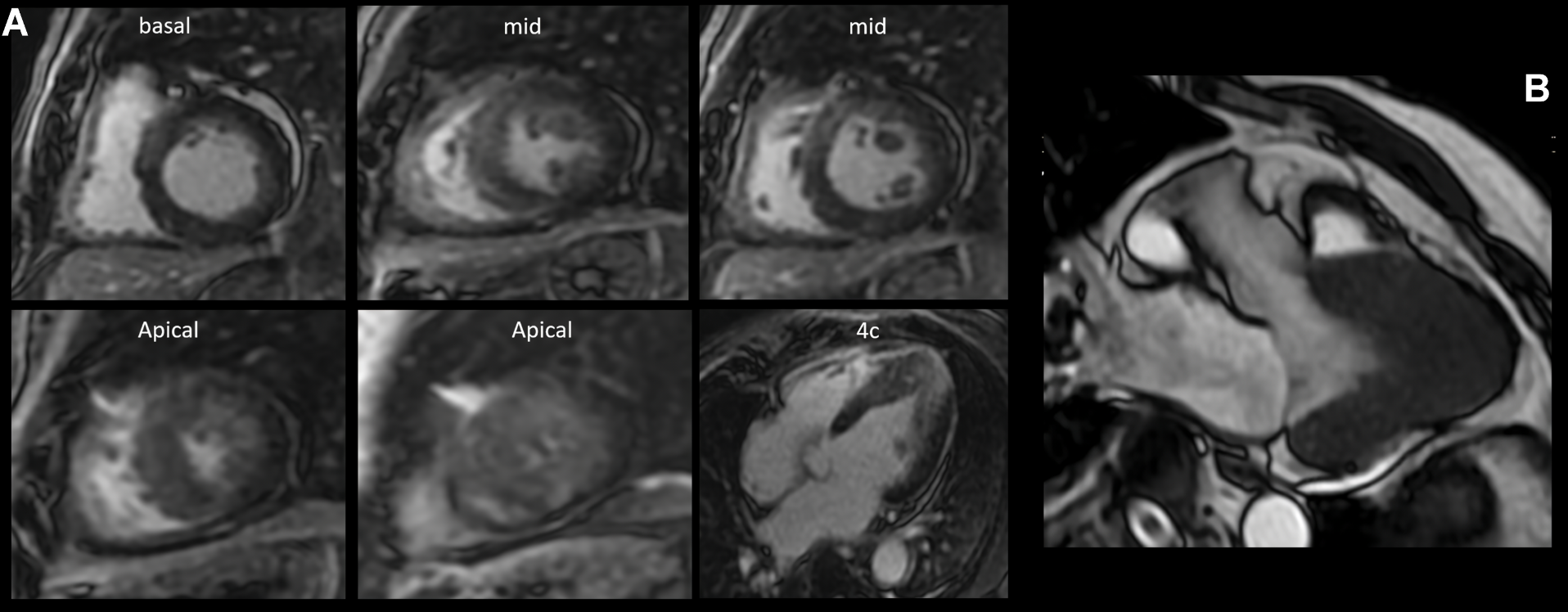
Figure 2. (A): Inversion-recovery sequence after intravenous contrast administration, enhancement was observed at 10 minutes. Basal third: intramyocardial septal. Middle third: Intramyocardial septal. Apical third: Intramyocardial in anterior, lateral, inferior and septal diffuse-patches. (B): Left ventricular 3-chamber view with a diastolic diameter of 50 mm, systolic diameter of 30 mm, Ace of Spades signs due to collapse in systole in the apical third of the LV. LV measurements at apical level with septal thickness of 18 mm, anterior wall 20 mm, lateral wall 23 mm and inferior wall 16 mm
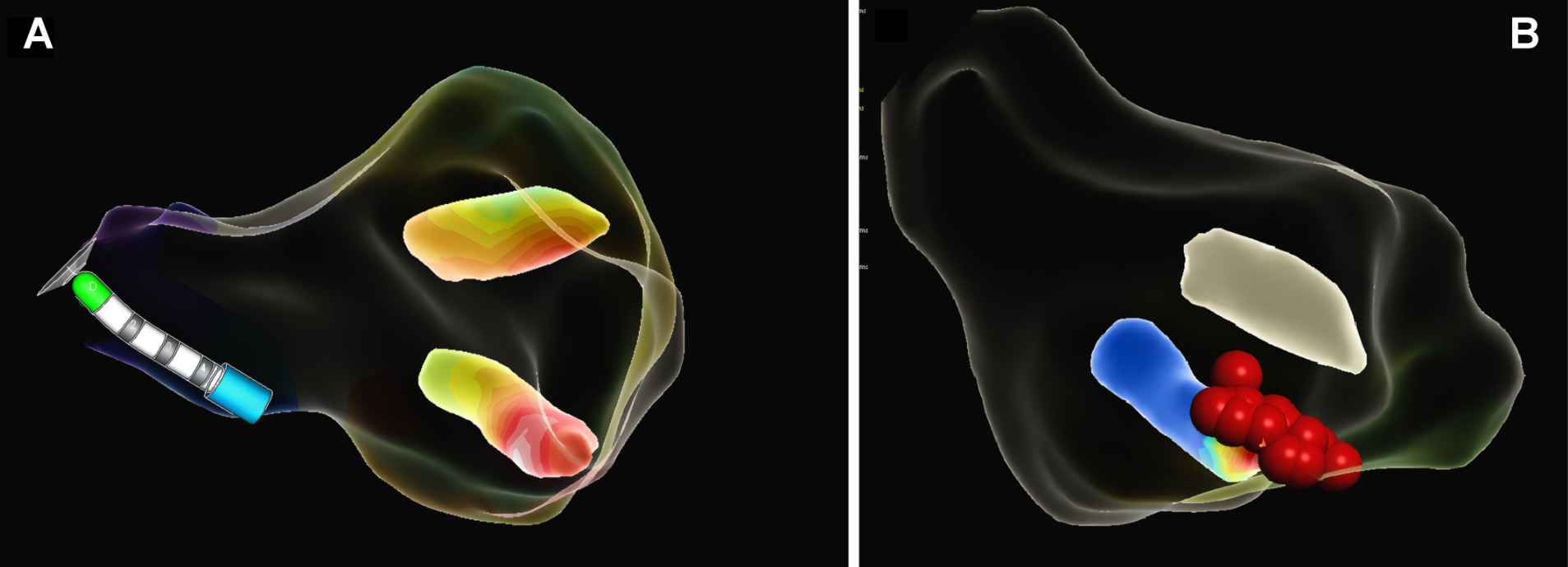
Figure 3. (A): Reconstruction of the three-dimensional electroanatomical mapping system identifying the focus of "focal monomorphic ventricular tachycardia" in the base of mitral posteroseptal papillary muscle. (B): Radiofrequency lesions in the posteroseptal mitral papillary muscle
Results and discussion
We present the case of a Latin American man diagnosed with apical hypertrophic cardiomyopathy, which is an uncommon subtype of HCM in Latin American subjects. This presentation is more common in Oriental individuals. AHCM is characterized by focal thickening of the apical myocardium of the left ventricle, showing a spade-shaped shadow or ace of spades in the left ventricle [2,7].
Regarding the clinical symptomatology of AHCM, there is no specific cardinal symptom. Dyspnea and chest pain stand out, which may be typical of angina pectoris; "palpitations" have also been described up to the occurrence of syncope [8]. Two-dimensional echocardiography is the initial study; however, MRI is the ideal study, since it allows obtaining high-resolution images to evaluate the different patterns of left ventricular thickening, especially when hypertrophy is limited to more distal segments [9]. The presence of asymmetric ventricular hypertrophy of the apex with apical thickness > 15 mm and a ratio between the maximum wall thickness and the posterior wall > 1.5 mm, according to echocardiography or MRI, electrocardiographic changes of left ventricular hypertrophy and negative precordial T waves in the absence of ischemic heart disease are elements to reach the diagnosis [10]. Some of these T waves are considered giant, being ≥ 10 mm, in addition to ST segment depression, prolonged QT has also been described [1,11].
This type of cardiomyopathy is characterized by multiple mutations that encode the proteins of the cardiac sarcomere and has a Mendelian range of expression with an autosomal dominant pattern of presentation. Acquired forms also exist [2,3,12]. Sarcomere protein gene mutations that cause apical hypertrophy rather than the more common morphologies of hypertrophic cardiomyopathy reflect interactions between genetic etiology, background modifier genes, and/or hemodynamic factors. Only a limited number of sarcomere gene defects (e.g., cardiac actin Glu101Lys) consistently produce AHCM [13]. In a cohort study of 1053 subjects with AHCM, only less than 10% had apical disease. This less common subtype was associated with a negative genetic test result up to 75% of the time. In contrast to previous publications suggesting a predilection for ACTC1/TPM1 mutations in patients with AHCM, the two most common genotypes (MYBPC3-HCM and MYH7-HCM) remained the most common among patients with a positive genetic test [14].
Regarding the prognosis of this pathology, Kyle Klarich, MD, director of the Mayo Clinic Cardiovascular Disease Fellowship in Rochester, Minnesota, conducted a review of Mayo Clinic's comprehensive echocardiographic databases that was published in 2013 in the American Journal of Cardiology. Klarich, et al. [15] and the colleagues retrospectively identified 2,662 individuals with hypertrophic cardiomyopathy evaluated at Mayo Clinic between June 1976 and September 2006. Of these individuals, 193 patients (7.3%) without confounding factors were classified as HCM, and follow-up was obtained in 187 of them (114 men, mean age 62 ± 19 years; 73 women, mean age 66 ± 16 years). The mean duration of follow-up was 94 ± 76 months. There were 55 deaths; 21 had non-cardiac causes, 27 were of unknown causes and 7 were of cardiac etiology. While Kaplan-Meier analysis demonstrated that the overall survival observed in this group of North American patients was significantly worse than expected, this finding was entirely due to excess mortality in women. Survival in men with AHCM was almost identical to that of age-matched controls. Multivariate predictors of higher mortality included female sex, age at first visit, chronic atrial fibrillation, and history of stroke. This study suggested that AHCM has different prognostic implications for affected women than for affected men [15]. Other studies in Japanese population have shown a more benign course of this disease [1,2,16], with the exception of elderly adults [17]. In another study during a follow-up of 13.6+/-8.3 years, cardiovascular mortality was found to be only 1.9% [10]. Interestingly, in non-Japanese population, forms of HCM, particularly the apical phenotype, had a less favorable prognosis [18]. Despite the light of evidence, ventricular tachyarrhythmia is known to be the main mechanism of sudden cardiac death in HCM, which is why risk markers for the primary prevention of sudden death in HCM have been described, including syncope or exercise-associated episodes, VT, massive left ventricular hypertrophy with a maximum wall thickness of 30 mm, family history of sudden death, and hypotensive blood pressure response during exercise. These criteria are extrapolated to the HCM variant when discussing the placement of an implantable cardioverter-defibrillator [19-22].
Moreover, in recent years predictors of additional arrhythmic events have been described, such as late gadolinium enhancement of the left ventricle > 15%, atrial fibrillation and the presence of LV apical aneurysm [8]. Contrary to prevailing concepts, the course of AHCM is not entirely benign and may be associated with clinical, electrocardiographic and arrhythmogenic abnormalities in the course of its natural long-term course [23]. Various working groups in AHF have detected, by Holter monitoring, the presence of symptomatic and asymptomatic non sustained VT in 18% and 5%, respectively. AF in 12%, and VF in 1%. Monomorphic VT occurs more often in the presence of aneurysms, possibly related to reentry around the aneurysm [24]. In our case, the typical electrocardiographic findings of Yamaguchi syndrome were present (nonischemic large negative T waves), and we were able to detect the sign of the "Ace of Spades" both in MRI and echocardiography. Apical compromise was also found in the strain technique [25]. During the electrophysiological study, focal VT was detected at the level of the postero-septal papillary muscle of the left ventricle. Sustained and non-sustained VT originating in the papillary muscles (Figure 3A – Figure 3B) are uncommon and are triggered by focal mechanisms that include increased cardiac automaticity or PVCs [26]. Treatment, in addition to genetic counseling and prevention of sudden death, consists of symptom control with the use of beta-blockers and non-dihydropyridine calcium antagonists [21]. Our patient received amiodarone after arrhythmic substrate ablation with improvement of symptomatology. Although the HCM risk-SCD was no higher than > 4%, Implantable Cardioverter Desfibrillator (ICD) placement as primary prevention was reasonable due to > 50% fibrosis in the apical segments, the presence of VT, and a maximum wall thickness of 23 mm. We emphasize that there are no predictive models applicable to AHCM, so the risk of this entity could be underestimated with conventional criteria for HCM.
Conclusion
Yamaguchi syndrome, a variant of hypertrophic cardiomyopathy, presents a diverse clinical spectrum, from relatively benign cases to life-threatening arrhythmias and sudden cardiac death. Early diagnosis and comprehensive management are essential to improve patient outcomes. This case underscores the importance of a multidisciplinary approach involving clinical cardiologists, imaging specialists, and electrophysiologists. Advanced imaging techniques, such as cardiac MRI and echocardiography, play a pivotal role in identifying the unique apical hypertrophy, while electrophysiology assessments are critical in stratifying arrhythmic risk. Genetic testing, though negative in our patient, remains a valuable tool for family screening and counseling, emphasizing that not all cases of apical HCM are linked to known mutations. Collaborative efforts across specialties are crucial to ensure personalized and optimized care for patients with this complex condition.
Acknowledgments
We appreciate the help of Dr. Edgar Martinez with the interpretation of the cardiac MRI.
Conflicts of interest
The authors declare no conflicts of interest.
References
- Sakamoto T, Tei C, Murayama M, Ichiyasu H, Hada Y, et al. (1976) Giant T wave inversion as a manifestation of asymmetrical apical hypertrophy (AAH) of the left ventricle echocardiographic and ultrasono-cardiotomographic study. Jpn Heart J 17: 611-629. [Crossref]
- Yamaguchi H, Ishimura T, Nishiyama S, Nagasaki F, Nakanishi S, et al. (1979) Hypertrophic nonobstructive cardiomyopathy with giant negative T waves (apical hypertrophy): ventriculographic and echocardiographic features in 30 patients. Am J Cardiol 44: 401-412. [Crossref]
- Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, et al. (2008) Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc 83: 630-638. [Crossref]
- Kitaoka H, Doi Y, Casey SA, Hitomi N, Furuno T, et al. (2003) Comparison of prevalence of apical hypertrophic cardiomyopathy in Japan and the United States. Am J Cardiol 92: 1183-1186. [Crossref]
- Abugroun A, Ahmed F, Vilchez D, Turaga L (2017) Apical hypertrophic cardiomyopathy: A case report. Cardiol Res 8: 265-268. [Crossref]
- López-Ponce de Leon JD, Estacio M, Giraldo N, Escalante M, Rodas Y, et al. (2023) Hypertrophic cardiomyopathy in a latin american center: A single center observational study. J Clin Med 12: 5682. [Crossref]
- Bedoya-Jaramillo T M, Arango-Moreno R, Duarte-Suárez NR, Duque-Ramírez, M (2022) Cardiomiopatía hipertrófica apical: Revisión de la literatura y presentación de caso. Revista colombiana de cardiologia, 28: 5.
- Steinberg C, Nadeau-Routhier C, André P, Philippon F, Sarrazin JF, et al. (2020) Ventricular arrhythmia in septal and apical hypertrophic cardiomyopathy: The French-canadian experience. Front Cardiovasc Med. 7: 548564. [Crossref]
- Klues HG, Schiffers A, Maron BJ (1995) Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J Am Coll Cardiol 26: 1699-1708. [Crossref]
- Eriksson MJ, Sonnenberg B, Woo A, Rakowski P, Parker TG, et al. (2002) Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 39: 638-645. [Crossref]
- Buitrago Gomez N, Herrera-Escandón A, Negrete-Salcedo A, Quiñones-Calvache C (2021) Miocardiopatía hipertrófica apical: Síndrome de Yamaguchi. Acta Medica Colombiana: AMC: Organo de La Asociacion Columbiana de Medicina Interna 46: 4.
- Arad M, Penas-Lado M, Monserrat L, Maron BJ, Sherrid M, et al. (2005) Gene mutations in apical hypertrophic cardiomyopathy. Circulation 112: 2805-2011. [Crossref]
- Castro Beiras A, Monserrat L, Hermida M (2003) Miocardiopatía dilatada familiar: Situación actual y beneficios clínicos de la investigación básica, Rev Esp Cardiol 56: 7-12.
- Tudurachi BS, Zăvoi A, Leonte A, Țăpoi L, Ureche C, et al. (2023) An update on MYBPC3 gene mutation in hypertrophic cardiomyopathy. Int J Mol Sci 24: 10510. [Crossref]
- Klarich KW, Jost CH, Binder J, Connolly HM, Scott CG, et al. (2013) Risk of death in long-term follow-up of patients with apical hypertrophic cardiomyopathy. Am J Cardiol 111: 1784-1791. [Crossref]
- Sakamoto T, Amano K, Hada Y, Tei C, Takenaka K, et al. (1986) Asymmetric apical hypertrophy: Ten years experience. Postgrad Med J 62: 567-570. [Crossref]
- Suganuma Y, Shinmura K, Hasegawa H, Tani M, Nakamura Y, et al. (1997) Clinical characteristics and cardiac events in elderly patients with apical hypertrophic cardiomyopathy. Nihon Ronen Igakkai Zasshi. 34: 474-481. [Crossref]
- Chikamori T, Mckenna WJ, Doi YL, Akizawa M, Yonezawa Y, et al. (1992) Comparison of clinical, morphological, and prognostic features in hypertrophic cardiomyopathy between Japanese and western patients. Clin Cardiol 15: 833-837. [Crossref]
- Maron BJ (2015) Historical perspectives on sudden deaths in young athletes with evolution over 35 years. Am J Cardiol 116: 1461-1468. [Crossref]
- Maron BJ, Haas TS, Ahluwalia A, Murphy CJ, Garberich RF, et al. (2016) Demographics and epidemiology of sudden deaths in young competitive athletes: From the United States National Registry. Am J Med 129: 1170-1177. [Crossref]
- Maron BJ, Maron MS (2013) Hypertrophic cardiomyopathy. Lancet 381: 242-255. [Crossref]
- Weissler-Snir A, Adler A, Williams L, Gruner C, Rakowski H, et al. (2017) Prevention of sudden death in hypertrophic cardiomyopathy: Bridging the gaps in knowledge. Eur Heart J 38: 1728-1737. [Crossref]
- Abinader EG, Sharif D, Shefer A, Naschitz J (2002) Novel insights into the natural history of apical hypertrophic cardiomyopathy during long-term follow-up. Isr Med Assoc 4: 166-169. [Crossref]
- Hughes RK, Knott KD, Malcolmson J, Augusto JB, Mohiddin SA, Kellman P, Moon JC, Captur G. Apical hypertrophic cardiomyopathy: The variant less known. J Am Heart Assoc 9: e015294. [Crossref]
- Forero-Saldarriaga S, Nieto-Zarate JA (2023) As de picas y miocardiopatía hipertrófica apical: Un signo para no olvidar. Arch Cardiol Méx 93: 109-111. [Crossref]
- Yamada T, Doppalapudi H, Mcelderry HT, Okada T, Murakami Y, et al. (2010) Idiopathic ventricular arrhythmias originating from the papillary muscles in the left ventricle: Prevalence, electrocardiographic and electrophysiological characteristics, and results of the radiofrequency catheter ablation. J Cardiovasc Electrophysiol 21: 62-69. [Crossref]