Abstract
Primary splenic angiosarcoma is an extremely rare malignancy with high metastatic potential and a very poor prognosis. We describe a rare case of splenomegaly in an 82-year old woman later on diagnosed as angiosarcoma at our institution.
Key words
spleen, angiosarcoma
Introduction
Primary splenic angiosarcoma is an extremely rare malignancy with high metastatic potential and a very poor prognosis. Splenic angiosarcoma was first described in 1879 by T. LangerhanS [1]. The clinical symptoms associated with splenic angiosarcoma are extremely variable and definitive diagnosis is always challenging and often late.
Case report
An 82-year old lady presented to the clinic with anaemia and weight loss. Investigations revealed low haemoglobin and an abdominal Computerised Tomography (CT) scan showed a 20cm nodular spleen, but no lymphadenopathy. Clinically splenic lymphoma was suspected. Bone marrow biopsy did not reveal any abnormality. In order to establish diagnosis, a splenectomy was performed.
The spleen measured 210 x 90 x 70 mm and weighed 1187 g. The splenic surface was nodular and showed subcapsular haemorrhagic/bluish nodular lesions (Figure 1A). The cut surface of the spleen was haemorrhagic and showed a multi-nodular appearance with nodules varying in size, ranging from 5 mm to 30 mm (Figure 1B).
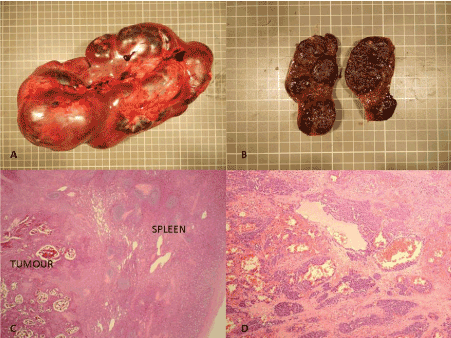
Figure 1. A-Macrophotograph of enlarged spleen showing multinodular architecture. B-Cut surface reveals haemorrhagic multinodular appearance. C-Microscopic section of spleen shows tumour with prominent vasoformative architecture (H & E 20X). D-Higher magnification showing tumour cells in islands separated by fibrous stroma (H & E 100X).
On microscopy, the splenic red pulp was partly replaced by a multi-nodular tumour demonstrating prominent vasoformative architecture (Figure 1C). Variably sized branching vessels were identified, which in places were lined by pleomorphic tumour cells. The nodular tumour islands were separated by thick fibrous septa (Figure 1D). The intervening stroma also contained solid sheets of tumour cells showing spindle to epithelioid morphology. These cells had round to oval pleomorphic nuclei with prominent nucleoli and a moderate amount of dense eosinophilic cytoplasm. Small vascular lumina with red blood cells were noted between the tumour cells (Figure 2A). The mitotic count was approximately 14 mitoses/ 10HPF. Areas of haemorrhage and necrosis were seen. There was no breach of splenic capsule and no evidence of any tumour nodules present outside of the capsule.
The tumour cells were positive for CD31 (Figure 2B) and cyclin D1 (Figure 2C) and negative for CD34, D2-40, EMA, AE1/AE3, S100, Melan-A and CD8. The proliferation index by MIB-1 immunostain was high (Figure 2D).
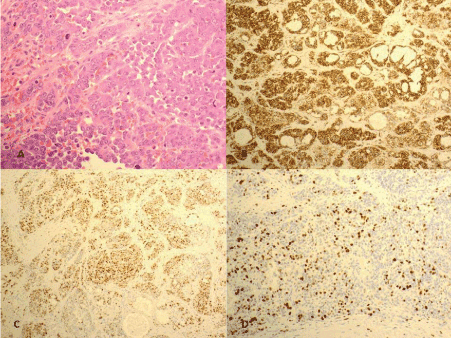
Figure 2. A-Epithelioid tumour cells showing round to oval pleomorphic nuclei with prominent nucleoli and a moderate amount of dense eosinophilic cytoplasm (H & E 400X). B-The tumour cells are diffusely positive for CD31 (100X). C- Nuclear expression of cyclinD1 is noted in tumour cells (100X). D-MIB-1 proliferation index is high (100X).
The post-operative recovery of our patient was uneventful. Unfortunately, a CT scan 3 months following surgery revealed widespread metastatic disease involving the liver and lungs. The patient was not fit for chemotherapy and best supportive care was offered in the community. The patient died 6 months after the surgery.
Discussion
Angiosarcoma of spleen is an extremely rare mesenchymal tumour of vascular endothelial origin with an annual incidence of 0.14-0.25 per million. Most of the patients affected are older; however, this disease may develop at any age. There is slight male preponderance, but no genetic predisposition has been identified. Although the pathogenesis of this tumour is not clear, exposure to ionizing radiation, arsenic, vinyl chloride and chemotherapy for lymphoma have been implicated as causative factors [1-4].
Clinical presentation of splenic angiosarcoma is variable with left upper abdominal pain being the most common symptom, seen in approximately 75% of patients in one series. Other complaints include fatigue, weight loss, and anorexia [3]. Splenomegaly is the most common physical examination finding and anaemia is the most common laboratory abnormality [1,4]. Definitive preoperative diagnosis of splenic angiosarcoma remains challenging. Due to risk of bleeding and malignant seeding, splenic biopsy is usually not an option for diagnosis. Hence, histology remains the gold standard for the diagnosis of this rare tumour which is possible only after splenectomy. Immunohistochemistry for vascular markers usually confirms the vascular origin of this tumour. Metastasis either at diagnosis or a few months after splenectomy is common and irrespective of treatment the median survival remains dismal at 5 months [4]. Neuhauser TS et al reported a mortality rate of 93% at 23 months [2].
In conclusion, this case highlights that splenic angiosarcoma is a rare highly aggressive tumour, that is difficult to diagnose clinically and may require surgical resection for definitive diagnosis. It may cause secondary spontaneous rupture, resulting in fatal haemorrhage. Since there is no established adjuvant therapy for the disease, median survival is poor even with adequate surgical treatment.
Author’s contributions
TB, SP and IB contributed to the conception and design of case presentation. TB collected the data and drafted the manuscript. SP performed the surgery and provided the clinical data. IB diagnosed the condition and revised the manuscript. All authors read and approved the final manuscript.
Consent for publication
Written informed consent was obtained.
Conflict of interest
The authors declare that they have no competing interests.
2021 Copyright OAT. All rights reserv
References
- Kamocki Z, Steward A, Zareba KP, Kuklinski A, Kedra B (2013) Primary splenic angiosarcoma –the same diagnosis yielding two different clinical pictures. Case report. Contemp Oncol (Pozn) 17: 218-221.
- Neuhauser TS, Derringer GA, Thompson LD, Fanburg-Smith JC, Miettinen M, et al. (2000) Splenic angiosarcoma: a clinicopathologic and immunophenotypic study of 28 cases. Mod Pathol 13: 978-987. [Crossref]
- Duan YF, Jiang Y, Wu CX, Zhu F (2013) Spontaneous rupture of primary splenic angiosarcoma: a case report and literature review. World J Surg Oncol 11: 53. [Crossref]
- Badiani R, Schaller G, Jain K, Swamy R, Gupta S (2013) Angiosarcoma of the spleen presenting as spontaneous splenic rupture: A rare case report and review of the literature. Int J Surg Case Rep 4: 765-767. [Crossref]