Abstract
An improved method using HPLC with diode array detection was developed for detecting isopropyl p-toluenesulfonate (IPTS) in palm based-isopropyl esters. The sample preparation includes an easy-to-handle solvent extraction technique which allows IPTS to be detected and quantified at very low levels. This method has a detection limit of 0.96 μg/g and a quantitation limit of 2.91 μg/g, respectively. The recovery of IPTS from two types of isopropyl esters, isopropyl palmitate, and isopropyl myristate, ranged from 96.1-102.1% and from 90.2-96.8 %, respectively. The coefficient of variation was less than 7%, demonstrating that the developed method is accurate and precise. The calibration curve (from 0.25 to 20 µg/ mL) has a correlation coefficient of 0.9999. This new method was used to analyse commercial isopropyl esters which were free from IPTS (based on analyses by the published GC-FID method). The results reconfirmed that IPTS was not detected in these commercial samples at 0.96 μg/g. A gas chromatography-mass spectrometer detector (GC-MSD) was also used to confirm the identity of IPTS in the spiked samples of isopropyl esters.
Keywords
oleochemical, genotoxic activity, isopropyl esters, isopropyl p-toluenesulfonate, HPLC, method validation
Introduction
Isopropyl esters are widely used in cosmetics and topical medicinal preparations, especially when the delivery of active ingredients through the skin is desired [1]. The traditional method used by many producers of isopropyl esters is the esterification of fatty acids with isopropyl alcohol. p-Toluenesulfonic acid is a common homogeneous catalyst used in this process [2]. During esterification, a side reaction termed as tosylation may occur. The process conditions (no base, anhydrous and high temperature) may induce tosylation. Tosylation involves the reaction of p-toluenesulfonic acid with isopropyl alcohol to form a by-product, isopropyl p-toluenesulfonate (IPTS).
The genotoxic activity of IPTS, or isopropyl tosylate, together with other types of alkyl tosylates, mesylates and besylates, has been indicated in an in vitro study [3]. To date, limits for genotoxic impurities are available only from a draft protocol proposed by the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA), mainly concerning drug substances and products [4,5]. So far, there has been no regulatory limit for IPTS in isopropyl esters. Chromatographic methods for the quantitative analyses of various types of sulfonate esters, including IPTS, have been reported but are limited to drug substances and products [6-14]. In our past work, the method using a GC-flame ionization detector (FID) for the determination of IPTS in palm oil-derived isopropyl esters was described. Using this method, the limit of detection was 12.5 µg/g and limit of quantification was 25 µg/g [15]. In this work, we report an improved method using an HPLC with a diode array detector (DAD). With this method, IPTS in palm oil-derived isopropyl esters can be detected and quantified with accuracy and precision at very low levels.
Experimental
Chemicals, materials and apparatus
AcidChem International Sdn Bhd, Pulau Pinang, Malaysia, provided the samples of isopropyl palmitate and isopropyl myristate used for the recovery studies. The isopropyl esters monitored for IPTS using the developed method were obtained from commercial companies. Apollo Scientific (Cheshire, UK) supplied isopropyl p-toluene sulfonate (97% purity). Both HPLC-grade acetonitrile (>99.0% purity) and purified water were supplied by Fischer Scientific (Pittsburgh, PA, USA). The Duran® volumetric flasks (10 mL) were from Schott Ltd (Mainz, West Germany), while the electronic dispenser, Multipette stream, with combitips of 0.2 and 2.5 mL were purchased from Eppendorf (Hamburg, Germany). The microvials (Part Number: 5182-0715) were obtained from Agilent Technologies (Palo Alto, CA, USA), and the vortex mixer was supplied by Benchmark Scientific Inc (New Jersey, USA). Hypersil Gold reverse phase C8 column (250 mm × 4.6 mm I.D, 5 μm particle size) was obtained from Thermo Scientific Ltd, (Walthan, MA, USA).
Calibration standard solutions
The initial standard stock solution of IPTS (≈ 1000 μg/mL) was prepared in HPLC-grade acetonitrile. It was then diluted using acetonitrile to 100 µg/mL as the secondary stock solution. Six calibration standards with concentrations ranging from 0.25 to 20 μg/mL were prepared by dilution of the secondary stock solution with acetonitrile.
Sample preparation for spiking/recovery study
In each case, isopropyl esters (2 g) were transferred to a 10-mL volumetric flask and spiked with IPTS using the initial standard stock solution (1000 μg/mL). Then, isopropyl esters containing three spiking levels of IPTS, namely 2.5, 5.0 and 50.0 µg/g, were prepared. IPTS was recovered from the spiked samples using a solvent extraction technique described here. Acetonitrile was filled to the mark (10 mL) and the mixture was homogenized for several minutes using a vortex shaker to facilitate extraction of IPTS into the acetonitrile fraction. After the mixtures settled into two layers, the upper tier (acetonitrile) was carefully transferred to another 10-mL volumetric flask until approximately 1 mL of residual acetonitrile remained above the ester layer. The second portion of acetonitrile (2 mL) was added, followed by a third portion (1 mL) for a second and third extraction, respectively. The collected acetonitrile extracts were then combined with the first extract. The combined extracts were adjusted accordingly to a volume of 10 mL in a volumetric flask prior to analysis with HPLC-DAD.
Sample preparation for monitoring IPTS in commercial isopropyl esters
The commercial isopropyl esters (2 g) were accurately transferred to a 10 mL volumetric flask. The samples were also spiked at 1 µg/g and 5 µg/g. The extraction process for IPTS from the isopropyl esters was the same as described for the spiking/recovery study. The 10 mL volumetric flask containing the combined extracts was filled to the mark with acetonitrile before HPLC-DAD analyses.
HPLC and analytical conditions
The analyses were performed using an Agilent Technologies HPLC (Palo Alto, CA, USA). The system was equipped with a diode array detector, a quaternary pump and a thermostated column compartment. Data acquisition and instrument control were performed using an Agilent Open Lab Chem Station software. The standard, spiked, and blank ester samples (5 µL) were injected into the HPLC system using an autosampler. Isocratic separation was performed on a reverse-phase C8 column. A mixture of acetonitrile:water (50:50 v/v) was used as the mobile phase. The flow rate was 1.0 mL/min and the column temperature was maintained at 24oC. External calibration was used for quantifying IPTS.
Identity confirmation by gas chromatography-mass spectrometry
The gas chromatography-mass spectrometer detector method (15) was adapted for detection of IPTS in a spiked isopropyl ester sample. The spiked sample was first analysed using the newly developed HPLC method, and the IPTS peak was identified by comparing it to the retention time of the IPTS standard. The same sample was then subject to GC-MSD analyses. The analyses were performed using a 7890A-5975C Agilent Technologies GC-MS fitted with an Agilent 19091J-413 MSHP-5 column (30 m length x 0.25 mm ID, 0.25 μm film thickness). The oven temperature was set at a starting temperature of 100ºC and maintained for 1 min, then ramped at 4ºC/min to 250ºC and kept at 250ºC for another 10 min. The samples (1 µL) were injected directly using a split mode of 10:1. An electron impact (EI) mode with a scan range of 35.0 to 500.0 was used. Helium (99.99% purity) was used as the carrier gas at a flow rate of 1.0 mL/min. Inlet temperature and pressure were 250ºC and 10.52 psi, respectively.
Results
The low level detection of IPTS in isopropyl esters e.g. isopropyl palmitate and isopropyl myristate was successfully developed using a HPLC-diode array detector. Figure 1 shows the representative HPLC chromatograms for IPTS standard (0.5 µg/mL), blank and isopropyl ester spiked at 2.5 µg/g of ester. The method was evaluated by performing system suitability test and method validation e.g. linearity, sensitivity measured as limit of detection and limit of quantification, precision and selectivity. The system suitability parameters obtained via analyses of an IPTS solution (1 µg/mL) showed the following: number of theoretical plates >2000, tailing factor <2.0, peak symmetry in the range of 0.9-1.2, and capacity factor of >2. The calibration curve has an equation of y = 127.39x + 3.1282 (Figure S1) and a coefficient of correlation (R2) of 0.9999. The detection and quantification limits for this method are 0.96 and 2.91 µg/g, respectively. Six repeated injections of an IPTS standard in acetonitrile at a concentration nearest to LOQ (0.5 µg/mL) were performed to evaluate the HPLC system precision. It was found that the injection precision for peak area was <2.96 %, while retention time was <0.8 %. Method precision was evaluated by preparing six independent samples of IPTS standard solution (2.5 µg/g). Each sample solution was injected once. The %RSD levels obtained were 4.21% (for IPM) and 9.53% (for IPP). Table 1 shows the accuracy results obtained from spiking/recovery of IPTS in isopropyl esters, IPP and IPM.
Table 1. Accuracy results obtained from spiking/recovery of IPTS in isopropyl esters, IPP and IPM.
Spiking level (μg g-1) | Mean recovery (%) ± SD (CV) n = 4 |
IPP | IPM |
2.5 | 102.1 ± 6.2 (6.08%) | 96.8 ± 4.2 (4.35%) |
5.0 | 96.5 ± 4.7 (4.92%) | 90.2 ± 3.9 (4.37%) |
25.0 | 96.1 ± 2.7 (2.84%) | 96.03 ± 2.8 (2.93%) |
IPP=isopropyl palmitate; IPM=isopropyl myristate; IPTS=isopropyl para-toluenesulfonate, SD = standard deviation, CV = coefficient of variation, n = number of replicates
Figure 2a shows a representative GC-MS total ion chromatogram (TIC) of a spiked isopropyl ester (25 µg/g). Retention time for IPTS in acetonitrile (5 µg/mL) using this method was 14.99 min (TIC not shown). The peak at 14.99 min in the spiked isopropyl ester (25 µg/g) was tentatively identified as IPTS. The peak in the spiked esters was further confirmed to be IPTS by using the NIST library available in the Agilent GC-MSD Mass Hunter software. The percentage match of the mass spectrum (at 14.99 min) from the spiked isopropyl esters samples (Figure 2b) and the library (Figure 2c) were 78.1% and 80.1% for IPM and IPP, respectively. This further confirms the identity of IPTS.
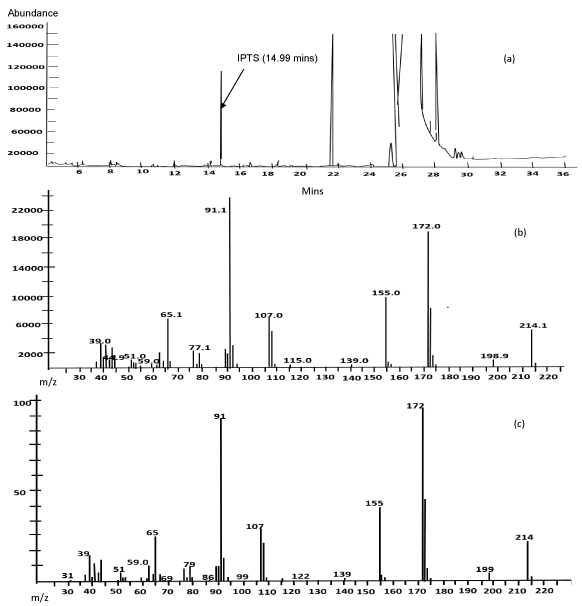
Figure 2. Total ion chromatogram (a) and mass spectra (b) of IPTS standard in IPP and mass spectra from the library (c).
Applicability of method to commercial isopropyl esters
The purpose of the study was to detect IPTS at a much lower detection limit than the reported GC-FID method, which was 12.5 µg/g (15). Eight types of commercial isopropyl esters from different sources were first analysed using GC-FID method, and IPTS was not detected. These same samples were again analysed using the newly developed method. The results showed that IPTS was also not detected at the much lower LOD of 0.96 µg/g. To further confirm the applicability of the new method, the same eight commercial samples were spiked at 1 µg/g (close to the detection limit), 5 µg/g and 25 µg/g. The method was found to be able to detect the IPTS peak spike at 1 µg/g in the commercial samples. The percentage recovery for all the eight spiked samples at 5 µg/g and 25 µg/g was within the acceptable limit of 80-120%.
Discussion
Sample preparation, chromatographic method development and system suitability
In previous work on analyses of IPTS in isopropyl esters by a GC-flame ionization detector, a simple sample preparation technique of dissolving and injecting was used prior to the GC analyses. Sample injection was performed using an inlet split ratio of 10:1 [15]. However, this sample preparation technique has some limitations. The presence of the matrix limits the amount of spiked IPTS able to be introduced into the GC inlet. If a much lower detection limit is required, it may be necessary for a higher sample volume to be introduced into the GC system. This was done by changing the split ratio or using a splitless inlet mode. However, the presence of the ester matrix may cause the GC column to become overloaded. This in turn may cause the matrix to interfere with the IPTS peak.
In this work, it was found that analysis of IPTS in isopropyl esters was possible using the HPLC-DAD method to detect IPTS in cosmetic matrices. However, the sample preparation and mobile phase composition were modified. The aqueous phase of the mobile phase mixture was HPLC-grade water without the addition of a buffer tetrabutyl ammonium phosphate, as in the method used for the cosmetic matrices [16]. The HPLC mobile phase mixtures was able to selectively detect IPTS in the isopropyl esters without the addition of a buffer. A different sample preparation technique was used for isopropyl esters. For cosmetic products, the first step was to dissolve the sample in acetonitrile, followed by homogenization using a vortex shaker. Then, each sample was filtered with a Teflon filter disc prior to injection into the HPLC system. However, the sample preparation technique used for cosmetic products was unsuitable for isopropyl esters. The esters were insoluble in acetonitrile and phase separation would occur, making direct injection impossible. IPTS from the esters was extracted using acetonitrile. IPTS had good solubility in acetonitrile but isopropyl esters were insoluble. Therefore, any IPTS present in the esters would be collected in the acetonitrile fraction during the mixing process. The direct injection method used for cosmetic products limits the amount able to be used during sample preparation. The method is only able to analyse IPTS without matrix interference using a weight/volume of 0.04 g/mL. In this work, the weight/volume used was 0.2 g/mL. The new limit of detection for IPTS in isopropyl esters (0.96 µg/g) was lower than in cosmetic matrices at 12.8 µg/g [16]. This modified HPLC condition used for cosmetic products was evaluated for its suitability in analyzing a different matrix such as isopropyl esters without matrix interference. Acetonitrile used as the extracting solvent during sample preparation was also used in the mobile phase mixture. The system suitability results further confirm the suitability of the developed conditions for the analyses of IPTS in palm oil-based isopropyl esters.
Method validation
The linearity of the method was evaluated using seven concentrations of IPTS standard solutions. The calibration curve was a plot of the mean responses measured as peak area for IPTS against concentration. The coefficient of correlation (R2) of 0.9999 for the calibration curve indicates good linearity (Figure S1).
The limits of detection (LOD) and quantification (LOQ) were calculated according to the formulae, 3.3 (Sy/S) and 10 (Sy/S), respectively. Sy is the standard deviation of the response of the curve, while S is the slope of the calibration curve. LOQ is typically chosen from the lowest concentration that can be measured with good accuracy and precision. In this study, the spiking level of (2.5 μg/g) closest to the calculated LOQ was chosen and evaluated accordingly. The results indicate that both accuracy and precision was good at the concentration of 2.5 μg/g as shown by a RSD of <7% for both types of isopropyl esters (Table I).
Spiking and recovery tests were carried out to evaluate the accuracy and precision of the new method. Percentage recovery was determined by spiking isopropyl palmitate (IPP) and isopropyl myristate (IPM) at four concentration levels. Isopropyl esters manufactured without the use of p-toluenesulfonic acid as a catalyst were used as spiking matrices. IPTS was extracted from the esters and analysed by HPLC-DAD. The recovered IPTS from the spiked samples was quantified using the external calibration curve. To ensure that the IPTS peak in the spiked sample was pure, the OpenLab ChemStation software was used to perform purity checks. In this work, it was found that the peak attributed to IPTS had no co-eluting impurity. Recovery of IPTS ranged from 96.1-102.1% in IPP and 90.2-96.8% in IPM (Table I), within the acceptable limits of 80 to 110% [4]. The intra-day precision of the recovery study measured by %RSD was <7%, showing good repeatability of the method. Percent recovery was calculated based on the results found in the spiked sample and the known added concentration. The blank isopropyl ester sample shows the absence of IPTS and other peaks from the matrix (Figure 1b). The chromatogram (Figure 1c) of a spiked IPP shows a peak at 12.3 min which was assigned to IPTS. This was based on a retention time comparison with an IPTS standard solution (Figure 1a). The precision of the HPLC system (for injection and retention time) and method precision were satisfactory as indicated by RSDs of less than 10 percent. The method was also found to have good selectivity as they can be applied for low level detection of IPTS in commercial isopropyl esters from different sources.
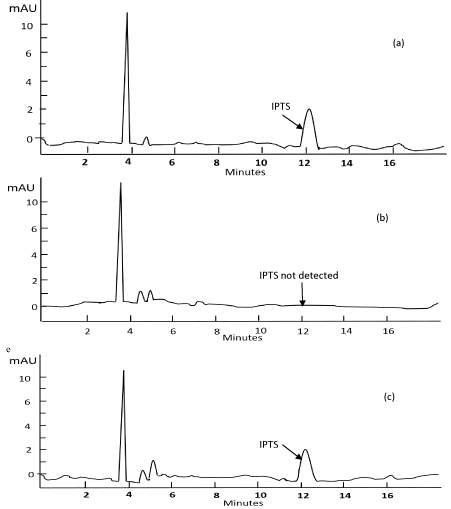
Figure 1. HPLC-DAD representative chromatograms of: (a) 0.5 μg ml-1 IPTS (b) blank IPP sample, and (c) spiked IPP at 2.5 μg g-1, monitored at 230 nm.
Conclusion
A rapid, simple, and improved method with the capacity for very low-level detection was developed using HPLC-DAD for detection and quantification of IPTS in isopropyl esters. The present method has a much lower detection limit than the GC-FID method [15]. The validation data demonstrates that the method is accurate, precise, sensitive, linear, and selective. Sample preparation is simple and easy to handle using a solvent extraction process.
Acknowledgement
The author wish to thank the Director General of MPOB for permission to publish this paper. This work is fully funded by Malaysian Palm Oil Board.
References
- Flick EW (1991) Cosmetics Additives: An Industrial Guide. Noyes Publication, Park Ridge, New Jersey.
- Sontag NOV (1979) Reaction of fats and fatty acids. In: Bailey’s Industrial oil and fat products (Swern, D., ed.), Wiley, New York, pp: 99-176.
- Glowienke S, Frieauff W, Allmendinger T, Martus HJ, Suter W, et al. (2005) Structure-activity considerations and in vitro approaches to assess the genotoxicity of 19 methane-, benzene- and toluenesulfonic acid esters. Mutat Res 581: 23-24. [Crossref]
- FDA (2008) Guidance for industry genotoxic and carcinogenic impurities in drug substances and products: United States Food and Drug Administration Recommended Approaches. http://www.fda.gov/cder/guidance/index.htm.
- EMA (2006) Guidelines on the limits of genotoxic impurities. Committee for Medicinal Products for Human Use, European Medicines Agency. http://www.emea.europa.eu
- Guo T, Shi Y, Zheng L, Zheng F, Feng F, et al. (2014) Rapid and simultaneous determination of sulfonate ester genotoxic impurities in drug substance by liquid chromatography coupled to tandem mass spectrometry: comparison of different ionization modes. J Chromatogr A 1355: 73-70. [Crossref]
- Devenport NA, Sealey LC, Alruways FH, Weston DJ, Reynolds JC, et al. (2013) Direct detection of a sulfonate ester genotoxic impurity by atmospheric-pressure thermal desorption–extractive electrospray–mass spectrometry. Anal Chem 85: 6224-6227. [Crossref]
- Wollein U, Schramek N (2012) Simultaneous determination of alkyl mesilates and alkyl besilates in finished drug products by direct injection GC/MS. Eur J Pharm Sci 45: 201-04. [Crossref]
- Kakadiya PR, Reddy BP, Singh V, Ganguly S, Chandrashekhar TG, et al. (2011) Low-level determinations of methyl methanesulfonate and ethyl methanesulfonate impurities in Lopinavir and Ritonavir Active pharmaceutical ingredients by LC/MS/MS using electrospray ionization. J Pharm Biomed Anal 55: 379-84. [Crossref]
- Sitaram C, Rupakula RB, Reddy BN, Sastry CSP (2011) Determination of alkyl methanesulfonates in doxazosin mesylate by gas chromatography-mass spectrometer. Indian J Pharm Sci 73: 107-110. [Crossref]
- Ramakrishna K, Raman NV, Rao KM, Prasad AV, Reddy KS (2008) Development and validation of a GC-MS method for the determination of methyl methanesulfonate and ethyl methane sulfonate in imatinib mesylate. J Pharm Biomed Anal 46:780-83. [Crossref]
- Taylor GE, Gosling M, Pearce A (2006) Low-level determination of p-toluenesulfonate and benzenesulfonate esters in drug substance by high-performance liquid chromatography/mass spectrometry. J Chromatogr A 1119: 231-237. [Crossref]
- Colon I, Richoll SM (2005) Determination of methyl and ethyl esters of methane sulfonic, benzene sulfonic and p-toluenesulfonic acids in active pharmaceutical ingredients by solid phase microextraction coupled to GC/SIM/MS. J Pharm Biomed Anal 39: 477-485. [Crossref]
- Li W (2004) Trace analysis of residual methyl methanesulfonate, ethyl methanesulfonate, and isopropyl methanesulfonate in pharmaceuticals by capillary gas chromatography with flame ionization detection. J Chromatogr A 1046: 297-301. [Crossref]
- Tay BYP (2013) Analysis of isopropyl para-toluenesulfonate in palm-based esters by gas chromatography with flame ionization detection and confirmed with mass spectrometry. Int J Cosmet Sci 35: 57-63. [Crossref]
- Tay BYP, Yung SC, Teoh TY (2016) Determination and confirmation of isopropyl p-toluenesulfonate in cosmetics by HPLC-diode array detector method and GC-MS. Int J Cosmet Sci 38: 627-33. [Crossref]