Alzheimer’s Disease (AD) is characterized by deposition of Amyloid-Beta Peptide (Aβ), which is produced from post-translational processing of Amyloid Precursor Protein (APP). The goal of this short review is to outline the major aspects of the biology of APP and the importance of APP in Alzheimer’s Disease. Subsequently, we briefly review current concepts regarding the potential targeting of APP processing in order to influence the ramifications of AD. Studies are underway to assess methods of altering the regulation of the APP gene as well as the regulation of the post-translational processing of APP. The complexity of APP processing has made the identification of new treatments for AD difficult. Future methods of targeting APP processing and expression are discussed.
Characteristics of the Most Common Type of Dementia: Alzheimer’s Disease
Post-translational modifications of Amyloid Precursor Protein (APP) are related to the Dementia disorder; Alzheimer’s Disease (AD) [1-3]. The incidence and pathophysiology of Dementia and Alzheimer’s Disease is important to consider when assessing genetic implications. Dementia is a broad category of many disorders that involve memory and cognitive impairment [1-3]. As the U.S. population continues to age, this problem will continue to increase as most types of dementia occur in later stages of life. Due to the immense physical, emotional, and financial toll that dementia can have on patients and their families, the importance of the development of therapies for this disorder continues to grow. Alzheimer’s Disease (AD) is the most common type of dementia with approximately 30 million patients currently affected by this disease around the world. It is estimated that 75% of all patients with dementia have AD [1]. The two main types of AD include early onset disease, which is generally caused by inherited genetic factors and late onset disease, which is hypothesized to be caused by a combination of genetic and environmental factors [1,4]. Early onset AD is rare, accounting for around 5% of cases of AD. As its name suggests, this type of AD displays symptoms in individuals at an earlier age than late onset AD. Early onset AD generally occurs during the fourth or fifth decade of life. Late onset AD is the more common form of AD and less likely to have identifiable genetic origins. Symptoms of late onset AD usually begin during the sixth or seventh decades of life. Many of the factors that contribute to late onset AD have yet to be determined and will be the focus of many ongoing research efforts [4]. The two primary pathophysiological hallmarks of Alzheimer’s disease are Neurofibrillary Tangles (NFTs) and β-amyloid (Aβ) plaques. These hallmarks have been studied since the early description of the disease by Alois Alzheimer in 1906 [5]. Although this review focuses on the formation of Aβ from APP, a brief introduction of NFT’s and Tau is warranted.
Neurofibrillary Tangles (NFTs) and Tau Protein in AD
NFTs are formed from hyperphosphorylated tau proteins, and they are known to form in the intraneuronal space in patients with AD [6]. Although APP has the strongest genetic link to AD, the Tau protein is also involved, and the interaction of APP and Tau remains the subject of much investigation. The Tau protein is known to facilitate the function and production of microtubules from tubulin. The Tau protein is associated with the formation of NFTs under conditions in which there is dysfunction of the kinases or phosphatases that interact with the Tau protein. This dysfunctional state leads to hyperphosphorylated Tau protein and the subsequent accumulation of tangles in the intraneuronal space [5]. Studies have also shown that the extent of NFT accumulation is correlated with the degree and duration of symptoms experienced by patients with AD [6,7]. Although there is clearly an association between the formation of NFTs and AD, the nature of this association is not clear as it has been shown that NFTs are not specific for just AD. NFTs are also present in other neurodegenerative diseases such as Parkinson’s Disease and posttraumatic dementia [6,8]. The fact that NFT’s are non-specific for AD is important. This finding has led to studies that are seeking to delineate the integral components that are specific to AD. Therefore, in more recent years, much attention has shifted to APP processing as a probable basis for AD. However, the interaction of APP and Tau remains an important area of investigation.
Characteristics of the APP Gene and APP
The APP gene encodes for Amyloid Precursor Protein. The APP gene is located on chromosome 21q21.3 and is approximately 240 kilobases in length (some variation in forms of APP). The location of the gene has been verified through studies performed on individuals with Down’s Syndrome. These studies show that the isoform of APP that is found in Aβ plaques is duplicated along with all other genes found on chromosome 21 in individuals with Down’s Syndrome. The early onset of AD in patients with Down’s Syndrome suggests a link of APP expression in AD pathology [4,9]. The genetic analyses of patients with AD have resulted in the discovery of many familial-associated mutations (FAM) of the APP gene that are believed to lead to the disease (10). These mutations occur in areas that encode for regions of the protein that will later result in increased Aβ production following post-translational processing of the APP protein [10]. Figure 1 outlines this process. It is noteworthy that genetic analyses of patients with AD have also revealed many mutations of the Presenilin genes (PS1 and PS2), which encode for the catalytic subunit of γ-secretase, an enzyme involved in the post-translational modification of APP [10-13]. The process of these post-translational modifications will be discussed in detail later. In mammals, the APP gene has two paralogs, APP like protein 1(APLP1) and APP like protein 2 (APLP2). The functions of these paralogs are mostly consistent in mammals [8,13]. The paralogs of the APP gene (APLP1 and APLP2) are unable to yield Aβ through post-translational processing. This characteristic is unique to the APP gene [8,14].
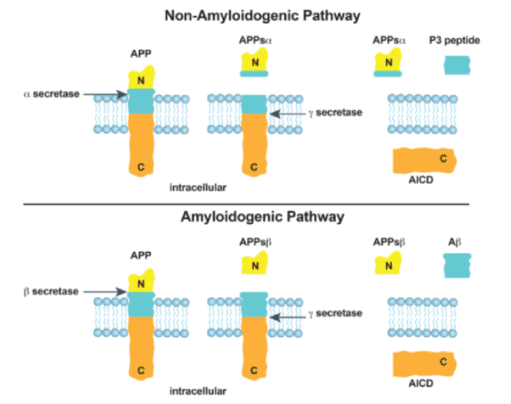
Figure 1. The two major pathways involved in post-translational processing of Amyloid Precursor Protein (APP) are depicted. Top: The non-amyloidogenic pathway utilizes the dual action of α-secretase and γ-secretase, which results in the truncated form of beta-amyloid (Aβ) known as P3. Bottom: The amyloidogenic pathway utilizes the dual action of β-secretase and γ-secretase to form Aβ
APP is a transmembrane protein, which has been linked to neurogenesis and the regulation of neural differentiation. There is a high level of expression of APP during brain development although APP has also been shown to be expressed in most tissues. Studies have shown that overexpression of APP causes differentiation, across different cell lines, to neuronal states [8,15-17]. Despite this evidence, investigators are still investigating the primary function of APP. In order to determine the function of APP and its complex relation to AD, it will be important to better understand the above noted mechanisms that modify APP. Additionally, the breadth of interactions of APP with other cellular events will require much study in the future.
Post-Translational Modifications of APP
For many years, it has been known that there are two major branches in the post-translational processing of APP. These two branches have been termed the amyloidogenic pathway and the non-amyloidogenic pathway (Figure 1), although recently, the simplicity of this terminology has been called into question as the two pathways may have important interactions in the development of Aβ and AD (3,18-21). These two major branches of post-translational modification involve three secretases: β-secretase, γ-secretase, and α-secretase (Figure 1). Again, recent reports have suggested that the simplicity of this system may be more complex with the recent discovery of additional secretases (η, δ, and alternative β-secretases) that interact with APP, and lead to metabolites that are the subject of current investigations (16,19). Even though this model of amyloidogenic/non-amyloidogenic APP processing may be more complex than originally believed, it has served as an important foundation for the understanding of the biology of APP processing and the components of Aβ in AD.
The first major pathway of post-translational APP processing, the amyloidogenic pathway, is depicted in Figure 1 [18,19]. This pathway begins as APP is cleaved by β-secretase to produce a soluble molecule, sAPPβ, that is secreted into the extracellular matrix and another molecule, βAPP-CTF, that remains bound to the membrane. The important difference between the two pathways, that are displayed in Figure 1, is the cut sites of β-secretase and α-secretase. The β-secretase cleaves APP at a location that does not interfere with the eventual formation of Aβ, while the α-secretase cleaves APP at a location that disrupts the formation of Aβ. The cut site of the β-secretase is essentially the first branch point in the difference between the amyloidogenic and non-amyloidogenic pathways of APP. βAPP-CTF is eventually cleaved by γ-secretase which results in the distribution of Aβ peptide into the extracellular matrix (Figure 1).
The second major pathway of post-translational APP processing, the non-amyloidogenic pathway, produces a P3 peptide (Figure 1). The P3 peptide represents a truncated form of Aβ (amyloid β-peptide 17-40/42). This pathway begins when the APP transmembrane protein is cleaved by α-secretase. The implementation of the cut site for α-secretase removes the possibility of Aβ production. After the protein is cleaved by α-secretase, a soluble sAPPα is secreted into the extracellular matrix and a αAPP C-terminal fragment (CTF) remains bound to the membrane. Next, a γ-secretase, cleaves the αAPP-CTF to produce a P3 peptide in the extracellular matrix and an amyloid precursor protein intercellular domain (AICD) inside the membrane.
These two post-translational pathways determine the possible functions of APP, and they are the subject of studies directed to reduce Aβ plaque formation. The interactions of the amyloidogenic and the non-amyloidogenic pathways are an area of much investigation. For instance, the P3 protein has been found to be present in Aβ plaques in patients with AD and Down’s Syndrome [15,20,21]. Lalowski, et al. [20] studied the brains of six patients with confirmed Down’s Syndrome. They examined pre-amyloid deposits in the cerebellums of these patients. They showed that the P3 peptide was a major component of pre-amyloid lesions. However, they demonstrated that the hydrophobic P3 peptide could form amyloid-like fibrils, but it could not form these fibrils as well as the full-length Aβ peptide that contained the N-terminal domain Aβ1-16 of the full-length APP, which inhibits exposure of the hydrophobic core. Further work will be necessary to delineate the role of the P3 peptide as it is found in the Aβ plaques. This work will involve seeking a better understanding of regulatory factors that are involved in pre-amyloid and Aβ formation as well as the mechanisms by which these processes progress.
When studying APP, it is important to explore studies that have examined the ramifications of APP knock-out. Knock-out studies of APP have been performed for more than 20 years. These studies have revealed much about the function of APP, and they have also demonstrated that APP has many interactions within the cell. Early studies generated knockout models with variations in methodologies. One of the primary methods that many researchers implemented to create these knockout models was the use of a targeting vector containing a disrupted APP gene and a drug resistance gene [22,23]. These targeting vectors were incorporated into embryonic stem cells. In a standard “knock-out” protocol, the stem cells were treated with the drug for which the targeting vector provided resistance in order to properly determine which stem cells incorporated the targeting vector through the process of homologous recombination [22,23]. After incorporating the disrupted APP gene, the stem cells were injected into mouse embryos at the blastocyst stage in order to generate APP knock-out models [22,23]
Early knock-out studies of APP provided many clues about APP and Aβ function. Several studies found that APP knock-out mice displayed decreased locomotor activity and also impaired neuronal function [22-24]. Dawson, et al. showed that APP knock-out mice demonstrated impaired long-term potentiation (LTP) in neurons, which is believed to be involved in memory formation through repeated neuronal responses [24]. This study focused on LTP processes in the hippocampus and found that the levels of gliosis of neuronal cells in the hippocampus were associated with deficits in LTP observed in APP knock-out mice. These findings suggested that APP likely plays a role in neuronal and synaptic development. Dawson’s findings suggested that Aβ has neurodegenerative effects that lead to synaptic loss. However, they also concluded that APP has neuroregenerative effects that may be useful to counteract the effects of Aβ under certain conditions. They hypothesized that the neuroregenerative properties of APP may be problematic in neurons that have dysfunctional neuritic sprouting, and that APP could increase neurodegeneration under certain conditions of increased Aβ. Because of this interplay with Aβ and APP, it is believed that there is a delicate balance that exists between the effects of these two proteins.
Another early APP knock-out study focused on how APP is involved in copper homeostasis in mice. APP and its different isoforms are known to contain binding sites for copper and zinc metals. Redox reactions involving these metals, in neuronal cells, can lead to the formation of harmful free radical molecules if these metals are not properly metabolized. White, et al. [25] made comparisons of the relative levels of these metals in different brain tissues between wild-type and APP knock-out mice. The results of this study showed that APP knock-out mice displayed significantly higher levels of these metals in the cerebral cortex. This finding suggested that APP likely plays an important role in the processing and clearance of potentially harmful metals in brain tissue [25].
The use of APP knock-out studies to further delineate the function of APP has continued to be the subject of investigation. As biotechnology continues to advance, tools such as CRISPR-Cas9 have become more useful in knock-out experiments [26]. CRISPR-Cas9 is a restriction endonuclease that can be used to create double strand breaks in targeted sequences of DNA. The specificity and ease of use of CRISPR-Cas9 technology has made it an increasingly common tool for researchers to use in knock-out studies (26). This technology has been used to target APP. In a study by György, et al. [27], an APP gene with a known familial Alzheimer’s Disease genetic mutation (Swedish) was targeted. CRISPR-Cas9 technology was utilized to knock-out this mutant APP allele both ex vivo and in vivo. Utilizing patient derived fibroblasts, CRISPR-mediated disruption of the mutant allele resulted in a substantial reduction in secreted Aβ. Additionally, in vivo studies were performed. An adeno-associated viral (AAV) vector was utilized to deliver CRISPR-Cas9 and disrupt the mutated allele in transgenic mice carrying the mutation. Following sacrifice of the animals and DNA analysis, the mutant alleles were found to be disrupted. The mutant alleles displayed indels whereas normal cerebellar tissue did not. These results suggest that future gene therapy approaches may be possible in familial-associated AD.
Mouse models that mimic Alzheimer’s Disease (AD) have undergone an interesting evolution during the last 25 years [28-30]. First generation knock-in models involved the knock-in of wild-type APP or mutated APP with known Familial Alzheimer’s Disease (FAD)-associated mutations of APP [30-38]. These various transgenic mouse models have been used for numerous studies of AD over the years. These first-generation models have been helpful to study various aspects of APP. For instance, these models have been helpful in the study of APP interactions during rigorous environmental manipulations and the progression of APP to Aβ. However, the first-generation models are generally associated with overexpression of APP and the possibility that non-physiological levels of APP may lead to misinterpretation of the results of these studies. Therefore, the results of these studies may be complicated by the production of APP fragments other than Aβ due to the high levels of APP.
Recently, investigators have sought to develop knock-in models of APP that are not associated with concomitant overexpression of APP. These new strategies constitute the second-generation APP knock-in strategies. Saito, et al. developed an APP knock-in strategy to accomplish this goal. This strategy of APP knock-in employed a method of humanizing the murine Aβ by changing three amino acids that differ between mice and humans (G676R, F681Y, and H684R) [39,40]. Additionally, they initially introduced two known FAD mutations. KM6701671: Swedish and 1716F: Beyreuther (Iberian) [40-44]. These APPNL-F mice were found to have increased Aβ42 and a high ratio of Aβ 42/ Aβ40, but they showed physiological APP levels. They also showed increased Aβ deposition in the cerebral cortex and hippocampus. This Aβ deposition was associated with memory dysfunction at an age of 18 months, as measured by the Y-maze test. Additionally, the mice demonstrated defects in special memory and reduced attention aptitude [39].
Next, APP knock-in mice with a third know FAD mutation were developed. This third mutation was the Artic mutation (E693G) and these AppNL-G-F mice also showed enhanced Aβ production. The AppNL-G-F mice demonstrated greater AD pathology and more significant cognitive dysfunction than the AppNL-F mice. These second-generation models have allowed for in vivo studies of AD with knock-in mouse models in which APP levels remain physiological. Both the first and second-generation knock-in mouse models display substantial differences in the phenotypic outcomes depending on the specific FAD-associated APP mutations [44]. Therefore, investigators have chosen a variety of FAD knock-in model systems to study APP in AD [42]. These APP knock-in systems have been useful for testing new cellular conditions or new therapeutics that may target APP-induced cognitive decline.
As stated above, many lessons have been learned from first-generation mouse models. They have been instrumental in foundational concepts of AD and remain useful today [30,36,45-47]. Early studies of male transgenic mice expressing the 751-amino acid isoform of human amyloid precursor protein (βAPP-751) demonstrated that the mice were less active and showed impaired spatial learning. However, histopathological evaluation of the mice did not show increased levels of Aβ deposition. These results suggested that increased levels of APP alone (without an associated increase in the accumulation of Aβ) may result in the compromise of neurological function under certain conditions [45]. During these early explorations of APP and Aβ deposition, it became evident that there was interplay between APP isoforms and other regulatory events. For instance, Quon, et al. explored the implications of the ratio of β-APP-751 (presence of Kunitz-type serine protease inhibitor) and β-APP-695 (absence of Kunitz-type serine protease inhibitor). They found that the increased ratio of β-APP-751/ β-APP-695 resulted in increased Aβ formation [46].
Another example in which the knock-in of an FAD-associated-mutated APP gene has been used to study the progression of AD is the investigation of the G-protein couple receptors; heterotrimeric guanine nucleotide-binding protein-couple receptor (GPCR) known as GPR3. GPR3 has been shown to be involved in γ-secretase activity and the generation of Aβ [48,49]. Therefore, Huang, et al. chose to study this association further by utilizing the APP/PSI transgenic mouse model (FAD: Swedish mutated APP with an additional mutation of Presenilin 1), which is known to demonstrate increased Aβ. They explored the effect of introducing deficiencies in one copy of GPR3. They found that Aβ levels were significantly decreased in the hippocampus of mice at 9 and 12 months, and pathological evaluation of the brains of these mice revealed significant reductions in amyloid plaque formation. Therefore, these results suggested that GPR3 activity may be a valuable target in the treatment of AD and may lead to marked reduction in amyloid plaques [50].
Therefore, these mouse models of AD have allowed investigators to gain an enormous amount of information regarding the mechanistic underpinnings of Aβ formation in AD. The biology of APP (see early summary) has demonstrated that there are potentially many aspects of APP expression and post-translational modification that can be targeted to reduce Aβ and amyloid plaque formation. Furthermore, there are many genes that influence various steps in Aβ metabolism and further delineation of this complex process will lead to new avenues of approaching this debilitating disease from a genetic basis. Other recent investigations of these processes are outlined in Table 1.
Table 1. Potential means to alter the ramifications of Alzheimer’s Disease-associated mutant APP
Model |
APP Manipulation |
Result |
Knock in mice: APP mutation and PS1 mutation [61]
|
Liqustilide, a lipophilic plant, that enhances alpha-processing of APP
|
¯ Memory impairment
¯ Aβ level
¯ Plaques |
Knock-in mice:
APPNL-GF mutation [62]
|
SAK3 treatment which is associated with release of acetylcholine, dopamine and serotonin
|
Previous studies have shown decreased dopaminergic and serotonergic pathways in APPNL-GF knock-in mice
|
Knock-in mice: APP Swedish mutant and PS1 mutant [63]
|
Resveratrol trimer, miyabenol C
isolated from stems and leaves of small-leaf grape plants
|
Inhibits β–Secretase and accumulation of β-Amyloid which may be mechanistically related to neuroprotection from Alzheimer’s Disease
|
Knock-in mice: APP Swedish mutant and PS1 mutant [64] |
Jujuboside A increases HSP90β - known stabilizer of proteins (suggest stabilization of proteins essential for Aβ clearance) |
¯ cognitive deficiency of mutated APP/PS1 transgenic mice
|
APP = Amyloid Precursor Protein; PS1= Presenilin 1; Aβ = Amyloid-Beta; SAK = Calcium Channel Enhancer
Many model systems are being used to study mechanisms of Aβ deposition that could eventually lead to treatments of AD. Many of these investigations are aimed at modifying post-translation processing of APP. In contrast to previously mentioned studies that explored methods to decrease γ-secretase activities, others have examined the significance of α-secretase. Explorations with α-secretase have centered on upregulation of α-secretase in order to decrease Aβ production (Figure 1). Upregulation of α-secretase leads to a soluble form of APP and potentially less formation of Aβ (non-amyloidogenic pathway). Therefore, in vitro studies were performed to examine the mechanism by which activation of the G-protein-coupled 5-hydroxytryptamine 4 (5-HT4) receptor leads to increased α-secretase activity and enhanced processing of APP to its soluble form [51]. These investigators found that 5-HT4-induced upregulation of α-secretase is dependent on Src-mediated activation of phospholipase C. These mechanistic steps may lead to improved methods of enhancing α-secretase processing of APP.
Recently, in vitro investigations of HEK293 cells with the FAD Swedish mutation of APP demonstrated that Bexarotene (a vitamin A derivative) interfered with Aβ accumulation and resulted in decreased Aβ levels [32]. However, these studies suggested that the reduction of Aβ was not due to the inhibition of γ-secretase cleavage as had been suggested by prior studies of Bexarotene. The authors hypothesized that Bexarotene may be a promising agent for reducing the accumulation of Aβ but may interfere with the toxicity of Aβ through mechanisms involving the decay of secreted Aβ. A better understanding of the mechanisms underlying these effects will contribute to future therapy-directed efforts.
Since autophagy has been implicated in neurodegeneration processes, the role of autophagy in AD has become the subject of investigation. Beclin 1 promotes autophagy and recruitment of membranes to form autophagosomes [52,53]. Immunoblots were performed to evaluate beclin 1 protein levels in the cells from the mid-frontal cortex gray matter of 8 patients who had died with moderate to severe AD. These beclin 1 levels were compared to levels in 11 non-demented control patients. The patients with AD were found to have beclin 1 levels that were 30% of controls. Interestingly, the beclin 1 levels in the gray matter of unaffected cerebellum in AD patients were comparable to the non-AD patients. It is hypothesized that beclin 1 deficiency disrupts lysosomes and promotes Aβ accumulation. Therefore, it is possible that upregulation of beclin 1 may be a potential therapeutic option in the future.
Finally, the significance of APP phosphorylation may be an important area of future investigation in AD. Recent investigations have shown that leucine-rich repeat kinase 2 (LRRK2), a gene in which mutations are common in Parkinson’s Disease (PD), phosphorylates APP at Thr668 and promotes enhanced transcription and accumulation of the APP intracellular domain (AICD). Also, these investigators found that this enhancement of AICD was associated with increased translocation of AICD to the nucleus. Further work regarding the significance of these events in patients with AD is warranted.
The above investigations of APP processing demonstrate that there are many pieces in this mechanistic puzzle. This complexity has made it difficult to translate laboratory finding into useful treatment approaches for patients. Future studies will be directed towards a better understanding of how these puzzle pieces are interconnected.
In this short review, the role of APP in AD has been presented. The processing of APP and the development of Aβ plaques is a complex sequence of events. We have discussed how investigators are seeking to manipulate these events at various junctures in the development of Aβ plaques. As we gain a better understanding regarding which events are critical in the path from APP to Aβ plaques, future studies may lead to the ability to alter these pathways at a genetic level. These future studies will involve an improved understanding of the regulation of critical proteins at the genetic level. The study of enhancer regions of critical proteins in the regulation of AD is an exciting new avenue of investigation [54,55].
Enhancer regions are cis-regulatory elements that increase the transcription levels of different genes. These elements are typically in the size range of 500 bp in length and can be bound by transcription factors that allow them to bind and interact with other regions of the genome [56]. These enhancer regions can be located as far away as 1 Mb from a region that is being enhanced. Enhancer regions typically interact with promoter regions. Promoter regions dictate where transcription of different genes will begin (Figure 2). The transcription factors of the promoters and enhancers interact, and the genomic region between the promoters and enhancers creates a “looping-in” event as depicted in Figure 2 [57,58]. There are many characteristics that indicate the presence of enhancer regions. Some of these characteristics include the histone marks, H3K27Ac, H3K4Me1, and H3K4Me3. DNase hypersensitivity and transcription factor binding sites are also considered to be markers for possible enhancer regions [56,57]. Due to the highly packed nature of chromatin and the fact that enhancer regions must be accessible to transcription factor proteins, all of these enhancer region marks are indicators of where the genome is “open” or accessible for these interactions [59]. As the name “histone marks” indicates, these interactions take place on histone proteins that have function in the coiling of DNA. H3K27Ac (acetylation of the 27th lysine of histone 3) and H3K4Me1 (mono-methylation of the 4th lysine of histone 3) are markers for active enhancers, while H3K4Me3 (tri-methylation of the 4th lysine of histone 3) is a marker for inactive enhancers. DNase hypersensitivity is used as a marker for possible enhancer sites as susceptibility to cleavage by DNase requires that the DNA is “open” and this is also a requirement for an enhancer region. Transcription factor marks are necessary marks for enhancer regions due to the nature of the interactions that take place between enhancer regions and other portions of the genome [56,57]. Through the use of the genome-wide Chromatin Immunoprecipitation (ChIP) assay, many of these different genomic characteristics have become accessible for the use of identifying possible enhancer regions [56].
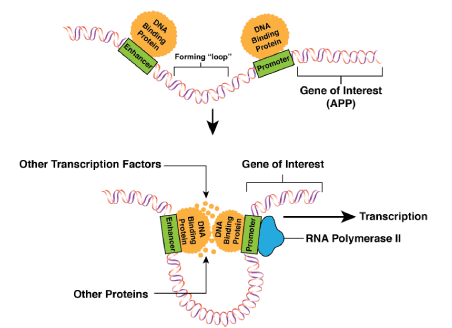
Figure 2. The three-dimensional interactions that take place between enhancer regions and promoter regions of the APP gene are depicted
Since enhancer regions are known to regulate the transcription of genes such as the APP gene and the genes that produce proteins that are involved in APP processing, they are an important subject of investigation regarding potential methods to reduce Aβ plaques. In the future, it will be important to develop therapeutics that have the potential to regulate the expression of the genes that are involved in APP expression and the genes that are involved in APP processing as well as Aβ plaque formation. The enhancer regions for these genes are potential targets for regulating these processes of the expression of these genes. Genetic databases and bioinformatics have created the ability to search for candidate genetic sequences that can subsequently be tested for this desired activity [60].
The author wishes to thank Dr. Richard Myers and Dr. Nicholas Cochran of the HudsonAlpha Institute for Biotechnology for education, encouragement, and direction in this research. This review represents a portion of an Auburn University Honors College independent research project.
- Chapman DP, Strine TW, Anda RF, Williams SM, Moore MJ (2006) Dementia and Its Implications for Public Health. Prev Chronic Dis 3: 1-13. [Crossref]
- Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA, Eldik JV (2014) Using Mice to Model Alzheimer’s Dementia: an Overview of the Clinical Disease and the Preclinical Behavioral Changes in 10 Mouse Models. Front Genet 5: 1-23. [Crossref]
- Murphy MP, Levine H (2010) Alzheimers Disease and the Amyloid-β Peptide. J Alzheimers 19: 311-323. [Crossref]
- Barber RC (2012) The Genetics of Alzheimer’s Disease. Scientifica 1-14. [Crossref]
- Kolarova M, García-Sierra F, Bartos A, Ricny J, Ripova D (2012) Structure and Pathology of Tau Protein in Alzheimer Disease. Int J Alzheimers Dis 1-13. [Crossref]
- Perl DP (2010) Neuropathology of Alzheimers Disease. Mt Sinai J Med 77: 32-42. [Crossref]
- Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, et al. (1995) Neocortical Neurofibrillary Tangles Correlate with Dementia Severity in Alzheimers Disease. Arch Neurol 52: 81-88.
- Nicolas M, Hassan BA (2014) Amyloid Precursor Protein and Neural Development. Development 141: 2543-2548. [Crossref]
- Tanzi R, Gusella J, Watkins P, Bruns G, George-Hyslop PS, et al. (1987) Amyloid Beta Protein Gene: CDNA, MRNA Distribution, and Genetic Linkage near the Alzheimer Locus. Science 235: 880-884. [Crossref]
- Zou Z, Liu C, Che C, Huang H (2014) Clinical Genetics of Alzheimer’s Disease. BioMed Research International 1-10. [Crossref]
- Nagata K, Takahashi M, Matsuba Y, Okuyama-Uchimura F, Sato K, et al. (2018) Generation of App Knock-in Mice Reveals Deletion Mutations Protective against Alzheimer’s Disease-like Pathology. Nat Commun 9: 1-7.
- Kelleher RJ, Shen J (2017) Presenilin-1 Mutations and Alzheimer’s Disease. Proc Natl Acad Sci 114: 629-631. [Crossref]
- Cai Y, An SS, Kim S (2015) Mutations in Presenilin 2 and Its Implications in Alzheimer’s Disease and Other Dementia-Associated Disorders. Clin Interv Aging pp:1163-1172. [Crossref]
- Fustiñana M, Ariel P, Federman N, Freudenthal R, Romano A (2010) Characterization of the Beta Amyloid Precursor Protein-like Gene in the Central Nervous System of the Crab Chasmagnathus. Expression during Memory Consolidation. BMC Neurosci 11: 1-11. [Crossref]
- Zheng H, Koo EH (2006) The Amyloid Precursor Protein: beyond Amyloid. Mol Neurodegener 1: 1-12. [Crossref]
- Freude KK, Penjwini M, Davis JL, Laferla FM, Blurton-Jones M (2011) Soluble Amyloid Precursor Protein Induces Rapid Neural Differentiation of Human Embryonic Stem Cells. J Biol Chem 286: 24264-24274. [Crossref]
- Chen CW, Boiteau R, Lai WF, Barger S, Cataldo A (2006) SAPP Enhances the Transdifferentiation of Adult Bone Marrow Progenitor Cells to Neuronal Phenotypes. Curr Alzheimer Res 3: 63-70. [Crossref]
- Andrew RJ, Kellett KAB, Thinakaran G, Hooper NM (2016) A Greek Tragedy: The Growing Complexity of Alzheimer Amyloid Precursor Protein Proteolysis. J Biol Chem 291: 19235-19244. [Crossref]
- Dulin F, Léveillé F, Ortega JB, Mornon JP, Buisson A, (2008) p3 Peptide, a Truncated Form of Aβ Devoid of Synaptotoxic Effect, Does Not Assemble into Soluble Oligomers. FEBS Letters 582: 1865-1870.
- Lalowski M, Golabek A, Lemere CA, Selkoe DJ, Wisniewski HM, et al. (1996) The “Nonamyloidogenic” p3 Fragment (Amyloid β17-42) Is a Major Constituent of Downs Syndrome Cerebellar Preamyloid. J Biol Chem 271: 33623-33631.
- Wang ZH, Gong K, Liu X, Zhang Z, Sun X, et al. (2018) C/EBPβ Regulates Delta-Secretase Expression and Mediates Pathogenesis in Mouse Models of Alzheimer’s Disease. Nat Commun 9: 1-16. [Crossref]
- Muller U, Cristina N, Li Z, Wolfer D, Lipp H, et al. (1994) Behavioral and Anatomical Deficits in Mice Homozygous for a Modified β-Amyloid Precursor Protein Gene. Cell 79: 755-765. [Crossref]
- Beglopoulos V, Shen J (2004) Gene-Targeting Technologies for the Study of Neurological Disorders. Neuromolecular Med 6: 13-30. [Crossref]
- Dawson G, Seabrook G, Zheng H, Smith D, Graham S, et al. (1999) Age-Related Cognitive Deficits, Impaired Long-Term Potentiation and Reduction in Synaptic Marker Density in Mice Lacking the β-Amyloid Precursor Protein. Neuroscience 90: 1-13. [Crossref]
- White AR, Reyes R, Mercer JF, Camakaris J, Zheng H, et al. (1999) Copper Levels Are Increased in the Cerebral Cortex and Liver of APP and APLP2 Knockout Mice. Brain Res 842: 439-444. [Crossref]
- Bonner AB (2018) CRISPR/Cas9 Gene Editing and Its Potential to Treat Common Diseases. Biomed J Sci & Tech Res 5: 1-6.
- György B, Lööv C, Zabrowski M, Takeda S, Kleinstiver B, et al. (2018) CRISPR/Cas9 Mediated Disruption of the Swedish APP Allele as a Therapeutic Approach for Early-Onset Alzheimer's Disease. Mol Ther Nucleic Acids 11: 429-440. [Crossref]
- Sakakibara Y, Sekiya M, Saito T, Saido TC, Iijima KM (2018) Cognitive and Emotional Alterations in App Knock-in Mouse Models of Aβ Amyloidosis. BMC Neurosci 19: 1-17. [Crossref]
- Balducci C, Forloni G (2010). APP Transgenic Mice: Their Use and Limitations. Neuromolecular Med 13: 117-137. [Crossref]
- Kitazawa M, Medeiros R, Laferla FM (2012) Transgenic Mouse Models of Alzheimer Disease: Developing a Better Model as a Tool for Therapeutic Interventions. Curr Pharm Des 18: 1131-1147. [Crossref]
- Citron M, Oltersdorf T, Haass C, Mcconlogue L, Hung AY, et al. (1992) Mutation of the β-Amyloid Precursor Protein in Familial Alzheimers Disease Increases β-Protein Production. Nature 360: 672-674. [Crossref]
- Kamp F, Scheidt HA, Winkler E, Basset G, Heinel H, et al. (2018) Bexarotene Binds to the Amyloid Precursor Protein Transmembrane Domain, Alters Its α-Helical Conformation, and Inhibits γ-Secretase Nonselectively in Liposomes. ACS Chem Neurosci 9: 1702-1713. [Crossref]
- Guardia-Laguarta C, Pera M, Clarimón J, Molinuevo JL, Sànchez-Valle R, et al. (2010) Clinical, Neuropathologic, and Biochemical Profile of the Amyloid Precursor Protein I716F Mutation. J Neuropathol Exp Neurol 69: 53-59. [Crossref]
- Lichtenthaler SF, Wang R, Grimm H, Uljon SN, Masters CL, et al. (1999) Mechanism of the Cleavage Specificity of Alzheimers Disease-Secretase Identified by Phenylalanine-Scanning Mutagenesis of the Transmembrane Domain of the Amyloid Precursor Protein. Proc Natl Acad Sci 96: 3053-3058. [Crossref]
- Tsubuki S, Takai Y, Saido,TC (2003) Dutch, Flemish, Italian, and Arctic Mutations of APP and Resistance of Aβ to Physiologically Relevant Proteolytic Degradation. Lancet 361: 1957-1958. [Crossref]
- Sakakibara Y, Sekiya M, Saito T, Saido TC, Iijima KM (2019) Amyloid-β Plaque Formation and Reactive Gliosis Are Required for Induction of Cognitive Deficits in App Knock-in Mouse Models of Alzheimer’s Disease. BMC Neurosci 20: 1-14. [Crossref]
- Nair RR, Corrochano S, Gasco S, Tibbit C, Thompson D, et al. (2019) Uses for Humanised Mouse Models in Precision Medicine for Neurodegenerative Disease. Mamm Genome pp:1-19. [Crossref]
- Lalonde R, Fukuchi K, Strazielle C (2012) APP Transgenic Mice for Modelling Behavioral and Psychological Symptoms of Dementia (BPSD). Neurosi Biobehav Rev 36: 1357-1375. [Crossref]
- Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, et al. (2017) APP Mouse Models for Alzheimers Disease Preclinical Studies. EMBO J 36: 2473-2487. [Crossref]
- Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, et al. (2014) Single App Knock-in Mouse Models of Alzheimer’s Disease. Nat Neurosci 17: 661-663. [Crossref]
- Pan X, Green BD (2019) Temporal Effects of Neuron-Specific Beta-Secretase 1 (BACE1) Knock-in on the Mouse Brain Metabolome: Implications for Alzheimer’s Disease. Neuroscience 397: 138-146. [Crossref]
- Gidyk DC, Deibel SH, Hong NS, McDonald RJ (2015) Barriers to Developing a Valid Rodent Model of Alzheimers Disease: from Behavioral Analysis to Etiological Mechanisms. Front Neurosci 9: 1-18. [Crossref]
- Oddo S, Caccamo A, Shepherd JD, Murphy M, Golde TE, et al. (2003) Triple-Transgenic Model of Alzheimers Disease with Plaques and Tangles. Neuron 39: 409-421. [Crossref]
- Onos KD, Uyar A, Keezer KJ, Jackson HM, Preuss C, et al. (2019) Enhancing Face Validity of Mouse Models of Alzheimer’s Disease with Natural Genetic Variation. PLoS Genet 15: 1-29. [Crossref]
- Dʼhooge R, Nagels G, Westland CE, Mucke L, Deyn PPD (1996). Spatial Learning Deficit in Mice Expressing Human 751-Amino Acid β-Amyloid Precursor Protein. Neuroreport 7: 2807-2812. [Crossref]
- Quon D, Wang Y, Catalano R, Scardina JM, Murakami K, Cordell B (1991) Formation of β-Amyloid Protein Deposits in Brains of Transgenic Mice. Nature 352: 239-241. [Crossref]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, et al. (2000) High-Level Neuronal Expression of Aβ1–42in Wild-Type Human Amyloid Protein Precursor Transgenic Mice: Synaptotoxicity without Plaque Formation. J Neurosci 20: 4050-4058. [Crossref]
- Thathiah A, Spittaels K, Hoffmann M, Staes M, Cohen A, et al. (2009) The Orphan G Protein-Coupled Receptor 3 Modulates Amyloid-Beta Peptide Generation in Neurons. Science 323: 946-951. [Crossref]
- Thathiah A, Horré K, Snellinx A, Vandewyer E, Huang Y, et al. (2012) β-Arrestin 2 Regulates Aβ Generation and γ-Secretase Activity in Alzheimers Disease. Nat Med 19: 43-49. [Crossref]
- Huang Y, Skwarek-Maruszewska A, Horré K, Vandewyer E, Wolfs L, et al. (2015) Loss of GPR3 Reduces the Amyloid Plaque Burden and Improves Memory in Alzheimer’s Disease Mouse Models. Sci Transl Med 7: 1-10. [Crossref]
- Pimenova AA, Thathiah A, Strooper BD, Tesseur I (2014) Regulation of Amyloid Precursor Protein Processing by Serotonin Signaling. PLoS One 9: 1-13. [Crossref]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, et al. (2008) The Autophagy-Related Protein Beclin 1 Shows Reduced Expression in Early Alzheimer Disease and Regulates Amyloid β Accumulation in Mice. J Clin Invest 118: 2190-2199. [Crossref]
- Chen ZC, Zhang W, Chua LL, Chai C, Li R, et al. (2017) Phosphorylation of Amyloid Precursor Protein by Mutant LRRK2 Promotes AICD Activity and Neurotoxicity in Parkinson’s Disease. Sci Signal 10: 1-11. [Crossref]
- Myrum C, Nikolaienko O, Bramham CR, Haavik J, Zayats T (2017) Implication of the APP Gene in Intellectual Abilities. J Alzheimers Dis 59: 723-735. [Crossref]
- Bonner D, Nguyen D, Cochran JN, Partridge C, Roberts B, et al. (2018) Mapping Enhancer Regions For Genes Associated With Neurodegenerative Diseases. Alzheimer’s & Dementia 14(7). Abstract no. 171. Presented at the Alzheimer’s Association International Conference. Chicago, IL.
- Sakabe N, Savic D, Nobrega MA (2012) Transcriptional Enhancers in Development and Disease. Genome Biol 13: 1-11. [Crossref]
- Zhu Y, Sun L, Chen Z, Whitaker JW, Wang T, et al. (2013) Predicting Enhancer Transcription and Activity from Chromatin Modifications. Nucleic Acids Res 41: 10032-10043. [Crossref]
- Mora A, Sandve GK, Gabrielsen OS, Eskeland R (2015) In the Loop: Promoter–Enhancer Interactions and Bioinformatics. Brief Bioinform 17: 980-985. [Crossref]
- Galimberti D, Scarpini E (2011) Behavioral Genetics of Neurodegenerative Disorders. Curr Top Behav Neurosci 12: 615-631. [Crossref]
- Buckley M, Gjyshi A, Mendoza-Fandiño G, Baskin R, Carvalho RS, et al. (2015) Enhancer Scanning to Locate Regulatory Regions in Genomic Loci. Nat Protoc 11: 46-60. [Crossref]
- Kuang X, Zhou HJ, Thorne AH, Chen XN, Li LJ, et al. (2017) Neuroprotective Effect of Ligustilide through Induction of α-Secretase Processing of Both APP and Klotho in a Mouse Model of Alzheimer’s Disease. Front Aging Neurosci 9: 1-13. [Crossref]
- Wang S, Yabuki Y, Matsuo K, Xu J, Izumi H, et al. (2019) Correction: T-Type Calcium Channel Enhancer SAK3 Promotes Dopamine and Serotonin Releases in the Hippocampus in Naive and Amyloid Precursor Protein Knock-in Mice. PLoS One 14: 1-18.
- Hu J, Lin T, Gao Y, Xu J, Jiang C, et al. (2015) The Resveratrol Trimer Miyabenol C Inhibits β-Secretase Activity and β-Amyloid Generation. PLoS One 10: 1-13. [Crossref]
- Zhang M, Qian C, Zheng ZG, Qian F, Wang Y, et al. (2018) Jujuboside A Promotes Aβ Clearance and Ameliorates Cognitive Deficiency in Alzheimers Disease through Activating Axl/HSP90/PPARγ Pathway. Theranostics 8: 4262-4278. [Crossref]