The hematopoietic transcription factor PU.1, a member of ETS (E26_Transformation_Spesific) family encoded by spleen focus-forming virüs proviral integration (SFPI1), is required for foetal [1] and adult hematopoiesis [2]. Function of PU.1 is essential during the development of myeloid [2,3] and B-cell lineage [3], and terminal differentiation of eosinophils [3]. PU.1 mutation results in a severe reduction in myeloid progenitors which can respond to multilineage cytokines [interleukin 3 (IL-3), IL-6, and stem cell factor (SCF)], but not to myeloid-spesific cytokines including granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage CSF (GM-CSF), and macrophage CSF (M-CSF) [3]. Given this complex interaction, PU.1 is likely to regulate the cell committent predominantly to granulocytic or monocytic. Additionally, PU.1/GFI1B axis has been shown to regulate hematopoiesis at the level of erytroid/myeloid commitment [4].
The complete inactivation of PU.1 activity resulted in loss of monocytic differentiation ability in a cell line model retrovirally transduced with PU.1-ERTM fusion gene. However, partial knock-down of PU.1 activity caused decreased binding capacity of histone acetylation genes. Wild type PU.1 induction restored both CCAAT/enhancer-binding protein α (CEBPα) binding sites and H3K27 acetylation [5].
In murine models, complete loss of PU.1 was found to be lethal [1]. However, reduced levels of PU.1 up to 80% in the bone marrow leads to the development of AML in mice within 3-8 months. Therefore, it was suggested that this critical PU.1 transcriptional activity threshold 20% is sufficient for the survival of myeloid progenitors, but not to sustain their further differentiation [6]. PU.1 mutations are uncommon in human AML, but altered PU.1 activity has been described in rare reports [4]. A somatic point mutation (D262N) in the erythroid-related transcriptional repressor GFI1B led to secondary AML in a patient initially diagnosed with myelodisplastic syndrome. There was a block in erythroid commitment and increased survival of myeloid progenitors due to SFPI1 repression. Intriguingly, this patient had not have this mutation before transformation of refractory anemia with excess blasts to AML [4].
Down-regulated PU.1 expression levels were also reported in AML with PML/RARα [7] and AML1-ETO fusion oncoproteins [8], or FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD) mutations (Figure 1) [9,10]. PML/RARα directly inhibits phosphatase and tensin homologue deleted on chromosome 10 (PTEN) expression via competing with PU.1. All-trans retinoic acid (ATRA) degradates PML/RARα which in turn results in increased levels of PU.1 and PTEN expressions [11]. AML1-ETO down-regulates PU.1 expression via binding PU.1 and displacing the co-activator c-jun from PU.1 [8]. Both expression and fuctional levels of PU.1 and CEBPα were suppressed by the FLT3 mutations [9]. Current therapeutic interventions include ATRA or FLT3 inhibitors (CEP-701) for overcoming these differentiation blockades (Figure 2).
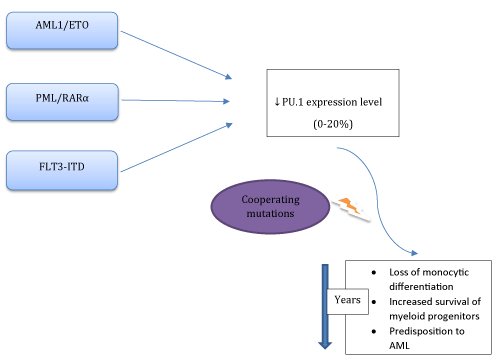
Figure 1. Abnormal fusion proteins (AML1/ETO, PML/RARα, FLT3-ITD) can alter PU.1 transcriptional activity via complex mechanisms. Reduction of PU.1 expression level below a critical threshold (<20%) and the additional cooperating mutations are associated with the accumulation of an undifferentiated myeloid progenitor pool and a predisposition to a preleukemic state.
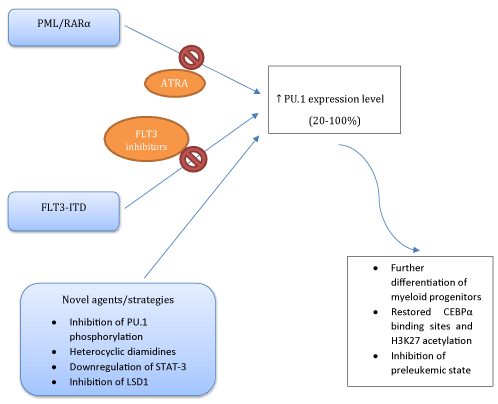
Figure 2. Current and novel therapeutic approaches to restore PU.1 activity and the results of increased PU.1 expression level
Different pharmacologic interventions are reported in order to restore the dysregulated PU.1 transcriptional activity. These efforts included reduced protein kinase C-δ (PKCδ)-mediated phosphorylation of PU.1 via IL-32θ [12], inhibition of the structure-dependent binding of PU.1 by heterocyclic diamidines [13,14], and honokiol induced downregulation of signal transducer and activator of transcription 3 (STAT-3) signalling via PU.1-induced SHPI1 activation [15]. Very recently, it has been shown that lysin spesific demethylase 1 (LSD1) inhibitor GSK-LSD1 ameliorated the expression of PU.1 [16]. Although these interventions are mainly experimental, they may provide an insight into the management of PU.1 dysregulated AML (Figure 2).
Taken together, these findings establish a causative link between PU.1 transcriptional activity and a normal myeloid differentiation. Additionally, dysregulated PU.1 expression levels are related with the pathogenesis of AML. Novel and targeted therapeutic approaches by ameliorating PU.1 expression levels seem to be promising. Future efforts are needed for a better understanding of PU.1 dysregulated AML and its individualized treatment.
References
- Scott EW, Simon MC, Anastasi J, Singh H (1994) Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science 265: 1573-1577. [Crossref]
- Staber PB, Zhang P, Ye M, Welner RS, Nombela-Arrieta C, et al. (2013) Sustained PU.1 levels balance cell-cycle regulators to prevent exhaustion of adult hematopoietic stem cells. Mol Cell 49: 934-46. [Crossref]
- Lee JJ, Rosenberg HF, eds. Eosinophils in Health and Disease. 1th ed.: Elsevier Inc., Academic Press; 2012. Maltby S, McNagny KM. Eosinophilopoiesis; chapter 5., p. 73-119.
- Anguita E, Gupta R, Olariu V, Valk PJ, Peterson C, et al. (2016) A somatic mutation of GFI1B identified in leukemia alters cell fate via a SPI1 (PU.1) centered genetic regulatory network. Dev Biol 411: 277-286. [Crossref]
- Sive JI, Basilico S, Hannah R, Kinston SJ, Calero-Nieto FJ, et al. (2016) Genome-scale definition of the transcriptional programme associated with compromised PU.1 activity in acute myeloid leukaemia. Leukemia 30: 14-23. [Crossref]
- Metcalf D, Dakic A, Mifsud S, Di Rago L, Wu L, et al. (2006) Inactivation of PU.1 in adult mice leads to the development of myeloid leukemia. Proc Natl Acad Sci U S A 103: 1486-1491. [Crossref]
- Walter MJ, Park JS, Ries RE, Lau SK, McLellan M,et al. (2005) Reduced PU.1 expression causes myeloid progenitor expansion and increased leukemia penetrance in mice expressing PML-RARalpha. Proc Natl Acad Sci USA 102: 12513-12518.
- Vangala RK, Heiss-Neumann MS, Rangatia JS, Singh SM, Schoch C, et al. (2003) The myeloid master regulator transcription factor PU.1 is inactivated by AML1-ETO in t(8;21) myeloid leukemia. Blood 101: 270-277. [Crossref]
- Mizuki M, Schwable J, Steur C, Choudhary C, Agrawal S, et al. (2003) Suppression of myeloid transcription factors and induction of STAT response genes by AMLspecific Flt3 mutations. Blood 101: 3164-3173. [Crossref]
- Zheng R, Friedman AD, Levis M, Li L, Weir EG, et al. (2004) Internal tandem duplication mutation of FLT3 blocks myeloid differentiation through suppression of C/EBPalpha expression. Blood 103:1883-90. [Crossref]
- Noguera NI, Piredda ML, Taulli R, Catalano G, Angelini G, et al. (2016) PML/RARa inhibits PTEN expression in hematopoietic cells by competing with PU.1 transcriptional activity. Oncotarget 7: 66386-66397. [Crossref]
- Kim MS, Kang JW, Lee DH, Bak Y, Park YS, et al. (2014) IL-32θ negatively regulates IL-1β production through its interaction with PKCδ and the inhibition of PU.1 phosphorylation. FEBS Lett 588: 2822-2829. [Crossref]
- Munde M, Wang S, Kumar A, Stephens CE, Farahat AA, et al. (2014) Structure-dependent inhibition of the ETS-family transcription factor PU.1 by novel heterocyclic diamidines. Nucleic Acids Res 2014;42(2):1379-90. doi: 10.1093/nar/gkt955. [Crossref]
- Stephens DC, Kim HM, Kumar A, Farahat AA, Boykin DW, et al. (2016) Pharmacologic efficacy of PU.1 inhibition by heterocyclic dications: a mechanistic analysis. Nucleic Acids Res 44: 4005-13. [Crossref]
- Bi L, Yu Z, Wu J, Yu K, Hong G, et al. (2015) Honokiol Inhibits Constitutive and Inducible STAT3 Signaling via PU.1-Induced SHP1 Expression in Acute Myeloid Leukemia Cells. Tohoku J Exp Med 237: 163-72. [Crossref]
- Cusan M, Cai SF, Mohammad HP, Krivtsov A, Chramiec A, et al. (2018) LSD1 inhibition exerts its anti-leukemic effect by recommissioning PU.1- and C/EBPa-dependent enhancers in AML. Blood 131: 1730-1742.