Background: Multiple factors have been associated with an increased risk of fetal growth restriction. The risk of genetic syndromes in these cases is not well established. The aim of this study was to determine the relationship between chromosomal abnormalities and early fetal growth restriction and to assess the incremental yield of genomic microarray over conventional karyotyping in fetuses with early growth restriction.
Methods: A prospective observational study of early fetal growth restriction diagnosed between 2013 and 2016 in our hospital. Chromosomal microarray analysis was performed in fetuses with early growth restriction defined as a fetal weight below the 3rd percentile estimated from 19 to 28 weeks of gestation, and a normal conventional karyotype result. We performed a descriptive analysis of the mean, the interval and the standard deviation for continuous variables and an analysis of absolute frequency and percentages for the categorical variables.
Results: Among 28 enrolled pregnant women, the incidence of early fetal growth restriction was 0.30%. We diagnosed 18.5 % of pathological results by arrays, but only one case (3.7%) were diagnosed by conventional karyotype too. Incremental yield of chromosomal microarray analysis over karyotyping was 16%. We detected a 20% incremental yield of chromosomal microarray analysis over karyotyping in fetus with structural anomalies, and a 17.6% incremental yield in isolated early fetal growth restriction.
Conclusions: The use of chromosomal microarray analysis provided a 16% incremental yield of detecting copy number variations in fetuses with early growth restriction and normal karyotype. Prenatal array should be part of the usual study of these fetuses, especially if there have ultrasound malformations associated.
chromosomal microarray analysis, array comparative genomic hybridization, genomic microarray, early fetal growth restriction, intrauterine growth restriction, prenatal diagnosis, copy number variation
Abbreviations
FGR: Fetal Growth Restriction, EFGR: Early Fetal Growth Restriction, CMA: Chromosomal Microarray Analysis, CNV: Copy Number Variations, SGA: Small for Gestational Age, EFW: Estimated Fetal Weight, VOUS: Variants of Unknown Significance
The conventional karyotype analysis has been the standard method for the detection of chromosomal abnormalities for decades. However, this technique is limited by the detection of chromosomal alterations that are bigger than 5-10 Mb. Sub-microscopic deletions and duplications, which are often associated with mental retardation and fetal malformations [1], are not usually detected with the karyotype. It is possible to evaluate the entire genome and detect copy number variations (CNVs), smaller than 50-60Kb, which equals an increase of 100 times the resolution compared with the conventional karyotype. Recent studies have focused specifically on the use of Chromosomal Microarray analysis (CMA) in prenatal diagnosis of fetuses with abnormal ultrasound findings [2]. When we reviewed the literature to evaluate the increasing diagnosis with microarrays in prenatal samples, we found that CMA detects 5.2% (IC 1.9 -13.9%) more cases of fetuses with structural malformation than the karyotype analysis [3,4]. Hence, few studies have reported detection rates for early fetal growth restriction (EFGR) fetuses.
The diagnosis of fetal growth restriction (FGR) is based on discrepancies between real and expected sonographic biometric measurements for determined gestational age. Traditionally, it has been defined as <10th percentile weight for gestational age and this establishes the diagnosis of small for gestational age (SGA). In our practice, when a fetus less than 10th percentile weight for gestational age is identified, we monitor fetal growth and fetal physiology over time. A normal growth evolution, normal Doppler velocimetry of the umbilical artery and normal amniotic fluid volume suggest a constitutionally small fetus or minimal fetal impact from uteroplacental insufficiency [5].
Multiple factors have been associated with an increased risk of FGR. It affects approximately 5%–10% of pregnancies and it is the second cause of perinatal mortality [6]. Various elements, including intrinsic fetal conditions as well as maternal, placental and environmental factors, can lead to intrauterine growth restriction. Some of these conditions have a genetic etiology: chromosomal abnormalities, monogenic diseases or epigenetic mutations. Up to 20% of FGR cases are attributed to a genetic cause [7-10].
Chromosomal abnormalities have been reported for up to 9.3-19% of fetuses with isolated FGR, and up to 4-21% in FGR with structural anomalies. Apparently, the CMA achieved a 4% incremental yield over karyotyping in non-malformed growth-restricted fetuses, and a 10% incremental yield in FGR with structural malformations associated [11-13]. Triploidies and trisomy 18 are the most common anomalies in fetuses before and after 28 weeks of gestation, respectively. However, the incidence of submicroscopic duplications/deletions and single-gene disorders with normal karyotype in less than 28 weeks of gestation, known as EFGR, is not well established.
We consider that it is important to establish the association between chromosomal aberrations and early fetal growth restriction (EFGR). By improving the diagnosis of these fetuses, we will improve the monitoring of the pregnancy, adapting the use of complementary examinations and, undoubtedly, providing the most accurate advice to families.
Case selection
This was a prospective observational study that included all the fetuses with less than 28 weeks of gestation affected with early growth restriction that were diagnosed in our hospital between 2013 and 2016. We analyzed: weeks of gestation when they were diagnosed, the use of additional diagnostic tests, fetal karyotype (conventional karyotype and chromosomal microarrays), associated malformations, how did the pregnancy end and the obstetric history.
The gestational age was calculated according to the last menstrual period and early pregnancy ultrasound results. All the ultrasound examinations were carried out by the authors using an Acuson Antares machine with a 2–3MHz convex transducer. The estimated fetal weight (EFW) was calculated from the biparietal diameter, abdominal circumference, and femur length using the Hadlock formula [14]. EFGR was diagnosed when the EFW was less than the 3th percentile for gestational age, in pregnancies with 28 weeks or less. The amniotic fluid volume and umbilical artery Doppler were assessed in all cases. EFGR was classified into two groups: isolated EFGR, and EFGR associated with others sonographic anomalies.
The cases included were those fetuses with a prenatal EFGR diagnosis defined as pregnancies below 28 weeks and weight below the 3th percentile. The indication for invasive prenatal diagnosis was having an EFGR. The exclusion criteria were the loss of gestational follow-up.
Chromosomal microarray analysis
The indication for invasive prenatal diagnosis was having an EFGR with or without sonographic abnormalities. Array comparative genomic hybridization compares the genomic content (DNA) of a patient (case) with a normal control and detects not only aneuploidy and major structural changes, but also submicroscopic gains or losses, and unbalanced reordering. Previously we needed to know if the samples came from a male or female fetus, for which the sample was subjected to a QFPCR (quantitative fluorescence polymerase chain reaction) to diagnose chromosomal abnormalities specific to whole chromosomes (chromosomes 13, 18, 21, X and Y), a process that can be automated and that allows lower costs and a faster diagnosis.
Amniotic fluid (20 mL) was collected by amniocentesis at 19–28 gestational weeks with informed consent. The analysis was carried out using an oligonucleotides microarray that compares genomic hybridization of approximately 60,000 probes distributed throughout the genome (qChip Pre v1.1 Complete, qgenomics). The DNA of the patient and internal reference DNA of the same sex with different fluorophores, Cy5 and Cy3 respectively, was marked. Subsequently the samples were hybridized on the array and scanned. The data obtained was analyzed using the Genomic Workbench 7.0 software. The average resolution of the array is 60kb, with higher resolution for areas of microdeletion-microduplication syndromes, telomeric and centromeric regions. The minimum number of consecutive oligonucleotides was established in five to detect an anomaly.
The variants identified were compared to those recorded in the Database of Genomic Variants (latest update May 2016). These variants were classified as pathogenic, VOUS (Variants of unknown significance, variants of uncertain clinical significance) or benign, following the recommendations of the American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants [15-18]. This report was positively rated by The European Molecular Genetics Quality Network.
Statistical analysis
The data was analyzed with SPSS 17.0. A descriptive analysis of the mean, the interval and the standard deviation for continuous variables, and of absolute frequency and percentages for the categorical variables was performed.
Incremental yield was calculated as the proportion of the abnormal results nondetectable by karyotyping divided by the total number of cases with an eventual normal karyotype [12].
This study was approved by the Institutional Ethics Committee of our hospital.
A total of 28 cases were detected. The incidence of early growth restriction in our studied population was 0.3%. The mean maternal age was 32.8 years, 40.7% were primiparous and 59.3% multiparous. The mean gestational age at diagnosis was 26.0 weeks in the patients examined. Other variables have also been analyzed (Figure 1).
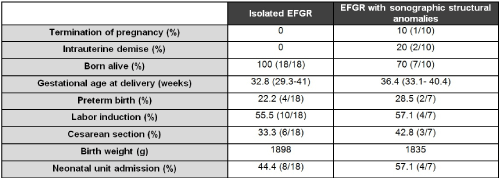
Figure 1. Pregnancy outcomes.
When we analyzed the obstetric history of the population studied, we observed that 7.4% of them had a prior history of a child with a neurodevelopment disorder and we had a case of a pregnant woman who have had a pregnancy with a fetus affected with trisomy 18. Only one case presented a recurrence of the diagnosis of fetal intrauterine growth restriction.
The 35.7%(10/28) of our cases had EFGR and sonographic alterations associated, 21.4% (6/28) of theme were structural fetal and 14.2% (4/28) non-structural alterations (Figure 2).
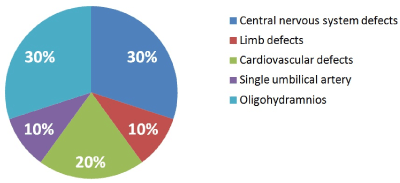
Figure 2. Malformations associated with EFGR.
Fetal MRI was performed in 17.8% of the cases (5/28), all of them affected with sonographic abnormalities, which were confirmed by the MRI in all the cases.
Fetal echocardiography was pathological in two cases: an apical muscular ventricular septal defect (VSD) and a right aortic arch.
The study of prenatal infections was negative in all cases, including those cases with mothers who were immune to Cytomegalovirus (CMV). In these cases, the amniotic fluid was studied by PCR-CMV without any pathological results in our series.
Fetal molecular karyotype and QF-PCR were performed in all of the fetuses diagnosed with EFGR, detecting one case of trisomy XXY (3.5%). Microarrays were conducted in all of the cases too and also allowed us to detect 5 cases of CNV (5/27,18.5%): 4 were variants of uncertain clinical significance (VOUS) and one was a pathogenic CNV. The cases of VOUS diagnosed were: a deletion of 203 kb in the chromosomal band 2q31.2, a duplication of 269Kb in the chromosomal band Xp22.31, a deletion of 1Mb in the chromosome band 5q35.3 and a deletion of 2.7 Mb in the chromosomal band 21q22.3, which did not match with polymorphic CNVs and could alter the structure or the reference gens. These alterations can be classified as VOUS since there is not enough literature that classifies them as benign or pathogenic. The pathogenic CNV detected was a duplication of 25Mb of chromosome bands 1q32.2q42.2, which was detected by conventional karyotype too, and altered the dose of more than 300 genes RefSeq, some of them described at OMIM (Online Mendelian Inheritance in Man) and clearly pathological (Figure 3).
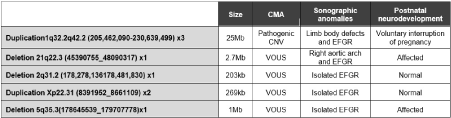
Figure 3. Detected CNV.
We diagnosed 18.5% (5/27) of the pathological results by arrays, but only one cases (3.7%) were diagnosed by conventional karyotype too. Incremental yield of CMA over karyotyping was 16%. We detected a 20% incremental yield of CMA over karyotyping in malformed growth-restricted fetuses, and a 17.6% incremental yield in isolated EFGR.
Legal interruption of pregnancy was requested by one of the patients, which represents a 3.5% of all the women studied. We had 2 cases of antenatal mortality (7.4%) and 48% (12/25) of the births were attended at the neonatal unit. Due to neurodevelopment problems, 28% (7/25) of the cases are in neurology follow-up at this moment.
When ultrasound examination suggests fetal growth restriction (FGR), prenatal care involves confirming the suspected diagnosis, determining the cause and severity of it, counseling the parents and determining the optimal time of delivery, if necessary. FGR resulting from intrinsic fetal factors, such as aneuploidies, congenital malformations or infections, often results in a prognosis that cannot be improved by any intervention.
The study of the natural history of the FGR has some peculiarities which make it difficult to establish its prevalence. On the one hand, defects in fetal growth are often left unidentified before birth. On the other hand, it doesn't help that scientific studies have generalized the use of small for gestational age (SGA) and FGR as synonyms. Small fetus by gestational age (SGA) refers to the fetus that simply has a weight below the expected threshold for weeks of pregnancy but that does not present a vital commitment and the Doppler velocimetry parameters are preserved, unlike what happens in the fetuses affected with growth retardation. All of the foregoing makes it difficult to establish a prevalence of FGR and even more when it comes to early FGR, where the data is less studied. It is described that the defect of fetal growth affects 5-10%13 of pregnancies, but how many of them have a delay in early fetal restriction growth is to be determined. Temming et al. [19] found that fetal growth restriction in the early second trimester occurred in less than 3% of the cases, but his study was retrospective. We have detected in our prospective serie a prevalence of only 0.3%, lower than the one reported by authors like Temming. We agree with Dall'Asta et al. [20] in the commentary on Temming, who defines the restriction of fetal growth as a fetal abdominal circumference lower than the 10th percentile and the pulsatility index of the umbilical artery greater than the 95th percentile. Recently, an international group of experts [21] in fetal medicine defined the early restriction of fetal growth as fetal biometric parameters lower than the 3rd percentile or a severely abnormal Doppler of the umbilically artery, final diastolic flow or two out of three of the biometrics below the 10th percentile and a pulsatility index of the umbilical or the uterine artery greater than the 95th percentile. Therefore, it is likely that most of the cases included by Temming et al. were fetuses constitutionally small and not with early restriction of growth, which are the ones pathologically small, hence his prevalence is higher than what we have found in our series.
Multiple factors have been associated with an increased risk of FGR. Genetic contribution to the causes of FGR has been established by 20% [11,12]. CMA provides high resolution genomic coverage by improving the diagnostic capacity of genetic studies. The incidence of submicroscopic duplications/deletions and single-gene disorders in FGR and normal karyotype is not well established. Karyotype analysis identified chromosomal aberrations in 9.3% of the cases, while CMA detected abnormalities in 18.8% of the cases. CMA achieved a 11.4% detection rate of chromosomal abnormalities among FGR cases with a normal karyotype [13]. Borrell et al. [12] reported results derived from this multicenter study that indicate that genomic microarray analysis leads to a 6.8% incremental yield over conventional karyotyping in fetuses with early growth restriction, and when stratified according to associated anomalies, this rate was 4.8% in isolated FGR and 10.5% in FGR with major structural anomalies. We detected from the analysis of our series that incremental yield of chromosomal microarray analysis over conventional karyotyping in fetuses with early growth restriction was 16%. We agree with both authors that chromosomal aberration is more frequent in the group of EFGR with associated anomalies. These results are in agreement with those published in the literature. Interestingly, the findings of our study demonstrate the added value of CMA compared with karyotyping, as it can be applied to not only evaluate FGR cases with sonographic anomalies but also FGR cases without anomalies.
Microarrays were conducted in all of the cases. We detected 5 cases of CNV (18.5%): four cases of VOUS and one case of a pathogenic CNV. One of the cases of VOUS diagnosed was a deletion of 203 kb in the chromosomal band 2q31.2. It does not match with CNV polymorphs and alters the structure and dose of the PDE11A gene (Phosphodiesterase 11A; OMIM 604961) [22]. Those specific mutations have been described as variants of predisposition associated with pigmented nodular adrenocortical hyperplasia and Cushing's disease.
The case of the double aortic arch was associated with a deletion in the chromosomal band 21q22.3, which altered the structure and the dose of 50 gens of reference, including COL6A1 and COL6A2 genes. These mutations have been associated with Bethlem myopathy with autosomal dominant inheritance pattern and Ullrich congenital muscular dystrophy [23,24], more severe and with recessive inheritance.
Another of the VOUS identified was a duplication of 269Kb in the chromosomal band Xp22.31, which also does not match with described polymorphic CNVs and could alter the structure or the dose of reference genes, such as KAL1 gene. The KAL1 gene encodes a protein, anosmin-1, that plays a key role in the migration of GNRH (OMIN 152760) neurons and olfactory nerves to the hypothalamus [25].
All the newborns affected with VOUS described previously presented an optimal postnatal evolution. However, in the case of the terminal deletion of the long arm of chromosome 5, the evolution was not so good. This deletion compromises the structure of 15 genes and 4 microRNAs. Among them, there are three morbid ones: ADAMTS2 (OMIN 604539) [26] associated to an autosomal recessive form of Ehlers-Danlos syndrome Type VIIC; LTC4S (OMIN 246530) associated to an autosomal recessive Leukotriene C4 synthase deficiency (OMIN 614037) [27]; and SQSTM1 (OMIN 601530) associated to an autosomal dominant Paget's disease (OMIN 602080) [28] and to an autosomal recessive type of neurodegeneration, ataxia and dystonia (OMIN 617145) [29]. Postnatally, this child developed a moderate ventriculomegaly during the first months of life and is under follow-up by neuropediatric. There is a possibility of requalifying VOUS as a pathogenic CNV if the scientific evidence suggests it.
The pathogenic CNV detected was a duplication of 25Mb of chromosome bands 1q32.2q42.2 (1q32.2q42.2(205,462,090-230,639,499) x3), which was detected by conventional karyotype. This duplication altered the dose of more than 300 genes RefSeq, some of them described at OMIM and clearly associated to pathology. Several pacifying duplications of chromosome 1 have been described in the literature and are associated with various phenotypes that include low stature, macrocephaly, low implantation ears, micro retrogression, heart defects, urogenital anomalies and intellectual disability. At the prenatal level, anomalies of chromosome 1 have been reported associated at the first trimester with an increased nuchal translucency and, in the second trimester, with anomalies as ventriculomegalies or omphaloceles, among others [30,31].
Regarding the perinatal results, it is difficult to find a population of study similar to ours, given the great diversity of studies and the poor homogeneity of themselves. In all the cohorts FGR pregnancies presented a risk for fetal death greater than for non-FGR pregnancies at all gestational ages. Story [32] carried out a study with the purpose of assessing the outcome of pregnancies where an estimated fetal weight less than the third percentile was detected prior to 24 weeks gestation. Twenty pregnancies were included in the analysis and 60% survived until discharge. Of the livebirths, 67% delivered preterm and 100% percent of livebirths were delivered by caesarean section. A prospective multicenter randomized study of fetal growth restriction management (Trial of Randomized Umbilical and Fetal Flow in Europe -TRUFFLE) [33] performed in 20 European perinatal centers between 2005 and 2010, included women with a singleton fetus at 26-32 weeks of gestation, with abdominal circumference < 10(th) percentile and umbilical artery Doppler pulsatility index > 95(th) percentile. The study showed that 2.4% of the babies died in utero, which was less than what was published by the Story [28] group.
According to the literature, FGR is a strong risk factor for stillbirth, neonatal death, preterm birth, ischemic encephalopathy, cerebral palsy, special educational needs and many other diseases in adult life [22,34,35]. In our study, we had a 7.4% rate of stillbirth. The survival rate was 92.6%, similar to those obtained by the TRUFFLE trial and superior to that presented by other authors as Story [27]. Few data exist on counseling and perinatal management of women after an antenatal diagnosis of early-onset fetal growth restriction. Moreover, the poor homogeneity in the works is still a major problem.
The chromosomal stud2021 Copyright OAT. All rights reservsed. Although our sample is small, our work shows that the application of the CMA in these cases contributes to a significant increased yield to the conventional karyotype, which entails better advice to the parents and an optimal control of the gestation.
Acknowledgements
The authors thank all women who enrolled in the study. They thank the principal investigators, collaborators and the staff members of the hospital for their contributions to the study.
Funding
This study was approved by the Institutional Ethics Committee of our center and supported by the Parc Taulí Research and Innovation Institute. The funders had no role in design of the study, data collection, analysis, interpretation of data or writing of the manuscript.
Availability of data and materials
The dataset supporting the conclusions of this article are available from the corresponding author on reasonable request.
Authors’ contributions
SP designed the study. JJ, LS, JL and NP acquired the data. JC analyzed the data. SP contributed to the conduct of the study. All reviewed and revised the manuscript, and approved the final manuscript as submitted.
Ethics approval and consent to participate
Written informed consent was obtained from all women prior to enrolment in the study. This study was conducted under the approval of the following institutional review boards or ethics committees: Ethical Committee on Clinical Research, University Hospital Parc Taulí, Sabadell, Barcelona, Spain.
Consent for publication
Not applicable. The present manuscript does not contain any individual person’s data in any form.
Competing interests
All other authors have no conflicts of interest to declare
- Hillman SC, McMullan DJ, Hall G, Togneri FS, James N, et al. (2013) Use of prenatal chromosomal microarray: prospective cohort study and systematic review and meta-analysis. Ultrasound Obstet Gynecol 4: 610-620. [Crossref]
- Tyreman M, Abbott KM, Willatt LR, Nash R, Lees C, et al. (2009) High resolution array analysis: diagnosing pregnancies with abnormal ultrasoun findings. J Med Genet 46: 531-541. [Crossref]
- Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, et al. (2012) Chromosomal Microarray versus Karyotyping for Prenatal Diagnosis.” The New England J Med 23: 2175-2184.
- Brady P (2014) A prospective study of the clinical utility of prenatal chromosomal microarray analysis in fetuses with ultrasound abnormalities and an exploration of a framework for reporting unclassified variants and risk factors. Genet Med 16: 469 -476. [Crossref]
- Unterscheider J, Daly S, Geary MP, Kennelly MM, McAuliffe FM, et al. (2014) Definition and management of fetal growth restriction: a survey of contemporary attitude. Eur J Obstet Gynecol Reprod Biol 2014; 174: 41-45. [Crossref]
- Lawn JE, Blencowe H, Pattinson R, Cousens S, Kumar R, et al. (2011) Stillbirths: Where? When? Why? How to make the data count? Lancet 377: 1448-1463.
- Kady S, Gardosi J (2004) Perinatal mortality and fetal growth restriction. Best Pract Res Clin Obstet Gynaecol 18: 397-410.
- Jacobsson B, Ahlin K, Francis A, Hagberg G, Hagberg H, et al. (2008) Cerebral palsy and restricted growth status at birth: population based case-control study. BJOG 115: 1250-1255. [Crossref]
- Gardosi J (2009) Intrauterine growth restriction: new standards for assessing adverse outcome. Best Pract Res Clin Obstet Gynaecol 3: 741-749. [Crossref]
- Gardosi J, Francis A (2009) Adverse pregnancy outcome and association with smallness for gestational age by customised and population based birthweight percentiles. Am J Obstet Gynecol 201: 28. e1-8. [Crossref]
- Snijders RJ, Sherrod C, Gosden CM, Nicolaides KH (1993) Fetal growth retardation: associated malformations and chromosomal abnormalities. Am J Obstet Gynecol 168: 547-555. [Crossref]
- Borrell A, Grande M.a, Pauta M.a, Rodriguez-Revenga L, Figueres F (2017) Chromosomal Microarray Analysis in Fetuses with Growth Restriction and Normal Karyotype: A Systematic Review and Meta-Analysis. Fetal Diagn Ther.
- Zhu H, Lin S, Huang L, He Z, Huang X, et al. (2016) Application of chromosomal microarray analysis in prenatal diagnosis of fetal growth restriction. Prenat Diagn 36: 686-692. [Crossref]
- Hadlock FP, Harrist RB, Sharman RS, Deter RL, Park SK (1985) Estimation of fetal weight with the use of head, body, and femur measurements-a prospective study. Am J Obstet Gynecol 151: 333-337. [Crossref]
- Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, et al. (2011) American college of medical genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 13: 680-685. [Crosrsef]
- Hanemaaijer NM, Sikkema-Raddatz B, van der Vries G, Dijkhuizen T, Hordijk R, et al. (2012) Practical guidelines for interpreting copy number gains detected by high-resolution array in routine diagnostics. Eur J Hum Genet 20: 161-165. [Crossref]
- de Leeuw N, Dijkhuizen T, Hehir-Kwa JY, Carter NP, Feuk L, et al.Diagnostic interpretation of array data using public databases and internet sources. Hum Mutat 33: 930-940. [Crossref].
- Jordan J, Simons A (2016) An International System for Human Cytogenetic Nomenclature. Cytogenetic and Genome Research 149: 1-2.
- Temming LA, Dicke JM, Stout MJ, Rampersad RM, Macones GA (2017) Early Second-Trimester Fetal Growth Restriction and Adverse Perinatal Outcomes. Obstet Gynecol 130: 865-869. [Crossref]
- DallʼAsta A, Lees C (2018) Early Second-Trimester Fetal Growth Restriction and Adverse Perinatal Outcomes. Obstet Gynecol 131: 739-740. [Crossref]
- Gordijn SJ, Beune IM, Thilaganathan B, Papageorghiou A, Baschat AA, et al. (2016) Consensus definition of fetal growth restriction: a Delphi procedure. Ultrasound Obstet Gynecol 48: 333-339. [Crossref]
- Horvath A, Boikos S, Giatzakis C, Robinson-White A, Groussin L, et al. (2006) A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nature Genet 38: 794-800. [Crossref]
- Park Y, Park MS, Sung DH, Sohn JY, Ki CS, et al. (2014) Ullrich Congenital Muscular Dystrophy Possibly Related with COL6A1 p. Gly302Arg Variant. Ann Rehabil Med 38: 292-296. [Crossref]
- Paco S, Casserras T, Rodríguez MA, Jou C, Puigdelloses M (2015) Transcriptome Analysis of Ullrich Congenital Muscular Dystrophy Fibroblasts Reveals a Disease Extracellular Matrix Signature and Key Molecular Regulators. PLoS One 10: e0145107. [Crossref]
- Cariboni A, Pimpinelli F, Colamarino S, Zaninetti R, Piccolella M, et al. (2004) The product of X-linked Kallmann's syndrome gene (KAL1) affects the migratory activity of gonadotropin-releasing hormone (GnRH)-producing neurons. Hum Mol Genet 13: 2781-2791. [Crossref]
- Colige A, Nuytinck L, Hausser I, van Essen AJ, Thiry M, et al. (2004) Novel types of mutation responsible for the dermatosparactic type of Ehlers-Danlos syndrome (type VIIC) and common polymorphisms in the ADAMTS2 gene. J Invest Derm 123: 656-663. [Crossref]
- Mayatepek E, Zelezny R, Lehmann WD, Hammond JW, Hoffmann GF (2000) Defects in the synthesis of cysteinyl leukotrienes: a new group of inborn errors of metabolism. J Inherit Metab Dis 23: 404-408, 2000. [Crossref]
- Laurin N, Brown JP, Lemainque A, Duchesne A, Huot D (2001) Paget disease of bone: mapping at loci at 5q35-qter and 5q31. Am J Hum Genet 69: 528-543. [Crossref]
- Haack TB, Ignatius E, Calvo-Garrido J, Iuso A, Isohanni P (2016) Absence of the autophagy adaptor SQSTM1/p62 causes childhood-onset neurodegeneration with ataxia, dystonia, and gaze palsy. Am J Hum Genet 99: 735-743. [Crossref]
- Kulikowski LD, Bellucco FT, Nogueira SI, Christofolini DM, Smith Mde A, et al. (2008) Pure duplication 1q41-qter: further delineation of trisomy 1q síndromes. Am J Med Genet A 146A: 2663-2667. [Crossref]
- Bocian M, Walker AP (1987) Lip pits and deletion 1q32-q41. Am J Med Genet 26: 437-443. [Crossref]
- Story L, Sankaran S, Mullins E, Tan S, Russell G, et al. (2015) Survival of pregnancies with small for gestational age detected before 24 weeks gestation. Eur J Obstet Gynecol Reprod Biol 188: 100-103.
- Lees C, Marlow N (2013) Perinatal morbidity and mortality in early-onset fetal growth restriction: cohort outcomes of the trial of randomized umbilical and fetal flow in Europe (TRUFFLE). Ultrasound Obstet Gynecol 42: 400-408. [Crossref]
- A Pilliod, YW Cheng, JM Snowden, AE Doss, AB Caughey (2012) The risk of intrauterine fetal death in the small-for-gestational-age fetus. Am J Obstet Gynecol 207: 318-416. [Crossref]
- Valcamonico A, Danti L, Frusca T, Soregaroli M, Zucca S, et al. (1994) Absent end-diastolic velocity in umbilical artery: risk of neonatal morbidity and brain damage. Am J Obstet Gynecol 170:796-801. [Crossref]