It is known that the sonic hedgehog (SHH) transcriptional activator GLI1 plays key roles in a panel of cancers including medulloblastoma (MB). We previously reported that AMP-activated protein kinase (AMPK) phosphorylates GLI1 and promotes GLI1 degradation through the β-TrCP-mediated ubiquitin proteasomal pathway. Therefore, activation of AMPK is a promising venue to target cancers that arise from aberrant SHH signaling such as MB. Mice models are commonly used to elucidate SHH-regulated developmental programs as well as SHH signaling in MB pathogenesis and would continue to inform how AMPK controls SHH signaling processes. However, an important question is whether AMPK regulates Gli1 in the murine system is not fully demonstrated. Here we review and present recent findings which demonstrate that AMPK downregulates Gli1 in mouse cell lines.
AMPK-activated protein kinase (AMPK), sonic hedgehog pathway, Gli1, metformin
The sonic hedgehog (SHH) pathway is a highly-conserved pathway important for cellular growth and differentiation as well as SHH signaling MB pathogenesis1,2]. During embryogenesis, spatial and temporal expression of SHH ligand coordinates the body patterning of the anterior-posterior as well as the proper orientation of limb buds [3]. In the central nervous system, SHH signaling directs the formation of the brain and spinal cord as well as growth and differentiation of glial cells and granule cell precursors [3-6]. SHH signaling plays critical roles in cerebellum development. The pattern of SHH expression was first mapped in mouse embryonic and post-embryonic development, revealing that the source of SHH are the Purkinje cells [7]. Mice harboring lacZ knock-ins at the Gli1 or Gli2 locus were later developed to follow the spatial and temporal pattern of SHH responses in cerebellar lobes and layers; robust expression of Gli1-lacZ and Gli2-lacZ was detected in the outermost external granule layer (EGL), where granule cells (GCs) proliferate [8]. However, high Gli1 expression was not detected in the inner EGL and internal granule layer (IGL) cells stained with NeuN, a marker for differentiating granule cells (GCs). Transgenic mice overexpressing SHH demonstrated normal cerebellar foliation but overall enlarged cerebellum and abnormal IGL [8]. Therefore, modifications of SHH signaling loci in transgenic mice in Corrales et al 2004 demonstrated that the spatial and temporal coordination of SHH signaling and Gli1 expression regulates both cellular and anatomical development of the cerebellum.
How SHH signaling regulates cellular growth and development, and how cells balance between a proliferative state versus differentiating state, are clinically important questions. Abnormal activation of SHH signaling results in various cancers such as basal cell carcinoma and medulloblastoma (MB), a highly aggressive cancer arising from the cerebellum. Numerous transgenic mice models have been developed for cancers involving mutations in SHH signaling components, including the PTCH1 heterozygous, SUFU heterozygous and p53 deletion, and SMOA2 mice [9-12]. Inhibition of GLI1 may correct aberrant SHH signaling that results in developmental defects or cancer, and much-needed novel targets or combinatorial targets can be revealed through understanding intersecting signaling pathways. Pharmacologic assessment of GLI1 downregulation in transgenic mice modeling MB may be ideal, but for these mice studies to be informative for human preclinical trials, they should be conducted with proof that the positive and negative regulation of SHH signaling is conserved in mice as they are in humans.
As cellular growth and development are both energy-consuming processes, the positive and negative regulation of SHH signaling is coordinated with cellular energy funds. A recent finding in Scientific Reports suggested a role for metabolic sensor AMP-activated kinase (AMPK) with SHH signaling-mediated cerebellar development. Liver kinase B1 (LKB1) is the primary kinase for AMPK. Mice harboring conditional knockouts of LKB1, mediated through Atoh1-Cre expression confined specifically to the cerebellar granule cell precursors (in addition to the inner ear, spinal cord, and intestines), were found to exhibit abnormal foliation concurrent with increased SHH target gene expression [13]. Thus, loss of LKB1, the primary AMPK kinase, results in increased SHH signaling, providing an indirect link between the metabolic sensor AMPK and SHH-mediated cerebellar development. In addition, the use of transgenic mice in the study once again underscores the importance of murine models in providing the groundwork for SHH signaling research.
Consistent with the Scientific Report findings, both our group and di Magno, et al. had previously revealed that AMPK phosphorylates GLI1 [14,15]. Phosphorylation by AMPK results in decreased GLI1 protein levels and activity, and a mutant cell line expressing GLI1 refractory to AMPK phosphorylation exhibits increased cellular proliferation and oncogenic potential [14,15]. Therefore, AMPK activation represents a promising venue for downregulating GLI1 protein levels and activity and may be assessed in preclinical studies as the next step. However, whether AMPK regulation of GLI1 is confined only to primates or is conserved in other higher eukaryotes such as mice has not been definitively demonstrated. Here we review evidence that AMPK regulates murine Gli1, providing the critical groundwork for mouse models of AMPK regulationof SHH signaling in cerebellar development and preclinical studies employing AMPK activation.
Cell culture
NIH3T3 cells and mouse embryonic fibroblast (MEF) cells were regularly passaged in DMEM supplemented with 10% bovine calf serum (BCS) and DMEM supplemented with 10% fetal bovine serum (FBS), respectively. Med1 cells were regularly passaged in DMEM supplemented with 10% FBS. For SHH signaling studies, NIH3T3 cells were serum-starved through growth in DMEM supplemented with 0.5% BCS. SHH stimulation was accomplished with SAG (100 nM) treatment for 24 hours. Pharmacologic AMPK activation was achieved with 25 uM A769662 for 24 hours (MTT assay) or two weeks (soft agar assay) or 25 mM 2DG for 1 to 3 days (MTT assay) or two weeks (soft agar assay), with medium replacement every two days.
Quantitative real-time PCR
Total RNA was isolated from NIH3T3 fibroblast cells using Trizol reagent (Invitrogen). One microgram of RNA was reverse-transcribed with random hexamer primers using SuperScript III reverse transcriptase (Invitrogen). A fraction (1/20) of the resultant cDNA was used as a template for amplification with TaqMan quantitative PCR probes (Applied Biosystems) on an Applied Biosystems 7500 Fast thermocycler: Gapdh (Mm99999915_g1), Gli1 (Mm00494645_m1).
Western blot analysis
MEF cells cultured in 0 mM, 5 mM, 10 mM, 15 mM, 20 mM and 25 mM glucose were lysed in lysis buffer. Total protein concentrations were determined using the Pierce BCA Protein Assay kit (ThermoFisher). Equal amounts of protein were resolved on a 10% SDS-PAGE gel, followed by transfer to a PDVF membrane after which the membrane was blocked in 5% milk. Antibodies were then diluted 1:1000 in blocking buffer for overnight incubation: Gli1 (Santa Cruz Biotechnology or Cell Signaling), p-ACC (Cell Signaling), AMPK (Cell Signaling), p38 (Cell Signaling) and Tubulin (Sigma).
MTT assay
5000 cells were plated in 96-well flat bottom plates and then exposed to either 5 mM (low) glucose, 25 mM (high) glucose or 25 mM glucose with 2DG. At the indicated times, 5 mg/ml MTT solution in phosphate-buffered saline (PBS) was added to each well for 1-4 hours. After removal of the medium, dimethyl sulfoxide (DMSO) was added to each well to dissolve the formazan crystals, and the absorbance at 570 nm was then determined. Triplicate wells were assayed for each condition.
AMPK activation suppresses Gli1 mRNA and protein expression
The application of AMPK activators (2DG, A769662 and AICAR) on NIH3T3 cells resulted in decreased Gli1 protein [14]. The same effect was observed in Med1, a mouse medulloblastoma cell line, and persists despite stimulation with SHH ligand [14]. We show in this report that NIH3T3 cells treated with AMPK-specific activator A769662 following SAG activation of the pathway resulted in decreased Gli1 activity, as exhibited through decreased Gli1 target gene transcript (Figure 1A). Consistent with these findings, di Magno et al. treated NIH3T3 cells with AICAR, 2DG or metformin (another AMPK activator), each of which suppressed SAG-induced Gli1 mRNA expression [15].
However, an important question is whether the Gli1 mRNA suppression is mediated by AMPK directly. In di Magno et al, AMPK knockout MEF cells were treated with AMPK activators AICAR, metformin or 2DG in the presence or absence of SAG (Figure 1B in [15]). In their experiments, the ability of AMPK activators to reduce Gli1 target gene expression persisted even in AMPK knockout MEFs, leading the authors to conclude that the inhibitory effect on Gli1 target gene expression in MEFs was independent of AMPK. In contrast to this experiment, we had previously examined Gli1 protein instead of target gene expression and discovered that while AMPK wild-type MEFs treated with 2DG inhibited Gli1 protein levels (Lane 2 vs Lane 1, Figure 1C in [14]), Gli1 protein levels remained the same in AMPK knockout MEFs under 2DG treatment (Lane 4 vs 3, Figure 1C in [14]). From these results, we concluded that AMPK activation resulted in a decrease of Gli1 protein, and that this decrease occurs in an AMPK-dependent manner in murine cells.
It is known that the activity of AMPK is controlled by the intracellular and intercellular glucose concentration, for example, AMPK activity is diminished in the diabetic patient, therefore AMPK agonists are clinically employed for the treatment of diabetes [16]. To examine whether Gli1 protein level is controlled by glucose through AMPK activity, AMPK wild-type and knockout MEFs were cultured in 0 to 25 mM glucose (Figure 1B). Consistently, Gli1 protein levels increased ~2.5-fold from 0 to 25 mM glucose and were inversely correlated to AMPK activity, as indicated by phosphorylated acetyl co-A carboxylase (p-ACC, a common AMPK substrate) (AMPK wt panels, Figure 1B). However, Gli1 protein levels remained relatively constant in AMPK knockout cells, suggesting that the effects on Gli1 protein levels in these experiments is AMPK-dependent (AMPK knockout panels, Figure 1B). Consistent with these results, the growth rate of AMPK wild-type cells increased in the high glucose compared to low glucose culture conditions, and this growth effect in high glucose cultures was reversed with 2DG activation of AMPK (Figure 1C). Together, these results demonstrate that activation of AMPK decreases Gli1 activity and protein levels in murine cultures.
That the AMPK regulation of Gli1 is not confined to primates is further supported from data in zebrafish as well as embryonic studies. Of note, supplementary data in the Li, et al. demonstrated AMPK regulation of Gli1 mRNA in zebrafish [14]. Zebrafish embryos treated with 2DG exhibited decreased Gli1 target genes transcription (Gli1, ptch1), while these same target gene mRNA levels increased after AMPKα1 antisense morpholino was injected (Figure S2 in [14]).
Mechanism of AMPK regulation of Gli1 in mice
The AMPK phosphorylation site(s) on Gli1 remain to be further elucidated. In the Cell Reports paper, we have laid groundwork towards this question [14]. Using mass-spectrometry, we detected three AMPK phosphorylation sites on GLI1: serine-408, serine-102 and threonine-1074 [14]. Serine-102 and threonine-1074 were not perfectly matched to AMPK consensus sites, however they were conserved in humans, mice and zebrafish. Serine-408 was detected only in human cells. However, mutation of serine-408 to alanine resulted in a 60% reduction of AMPK phosphorylation on GLI1, while mutation of all three sites resulted in the complete loss of AMPK phosphorylation. This result suggests that threonine-1074 and serine-102 are possible AMPK phosphorylation sites in both human and murine Gli1 and provide a possible direct mechanism of AMPK regulation of Gli1 in mice.
In addition, it was shown that β-TrCP-mediated GLI1 ubiquitination occurs following AMPK activation and results in GLI1 protein degradation [17]. This regulation is present in both human and murine cell lines. In mouse cells, Gli1 exhibited decreased protein levels upon 2DG treatment in a dose-dependent manner, which was accompanied by increased phospho-AMPK levels (Figure 4A in [17]). However, the Gli1 protein decrease was not observed in β-TrCP knockout cell lines (β-TrCP-/- panels, Figure 4A in [17]), indicating that β-TrCP is required for AMPK-induced Gli1 degradation in the mouse cell line.
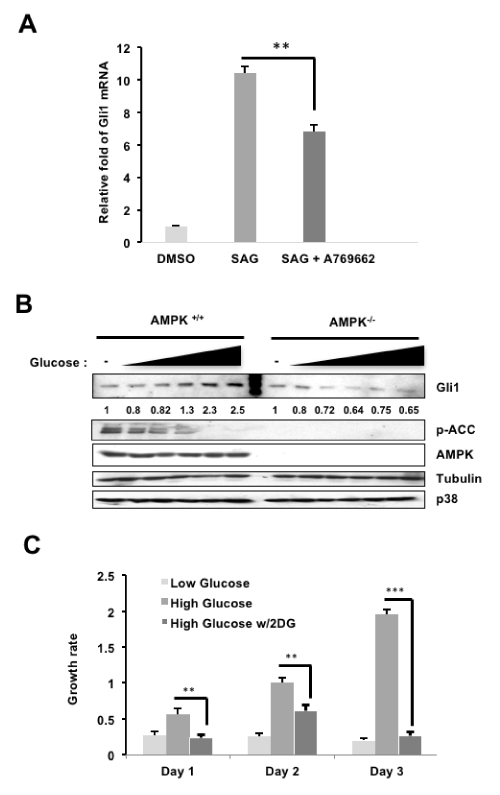
Figure 1. AMPK regulation of Gli1 in murine cells (A: NIH3T3 cells were serum-starved (SS) in DMEM (0.5% BCS) overnight, stimulated with SAG with or without A769662 (25μM) for 24 hours. The amount of Gli1 RNA was then analyzed by qRT-PCR and normalized to Gli1 mRNA levels from the DMSO group. B: AMPK wild-type or knockout MEF cells were cultured in increasing concentrations of glucose (0-25mM), and lysates were harvested and subjected to western blots in which Gli1, p-ACC, and AMPK protein levels were examined. C: MEFs cultured in 5 mM or "low" glucose, 25 mM or "high" glucose, or 25 mM "high" glucose with 25Mm 2DG treatment were subjected to MTT cell growth assays over three days)
Activation of AMPK inhibits MB growth
The previous results showed that activity of AMPK can be modulated by glucose leading to altered cellular growth. For example, inhibition of AMPK (high glucose) and AMPK activation (low glucose) can promote and reduce cell growth, respectively in MEFs (Figure 1C). We further investigated this result on the murine MB cell line Med1, a SHH pathway-driven MB derived from Ptch1-/+ mouse [16]. We cultured Med1 cells in either 5 mM "low" glucose, 25 mM "high" glucose or 25 mM "high" glucose with the AMPK activator 2DG. At day 2 and day 3 of culturing, Med1 cells cultured in AMPK inhibition conditions (high glucose) exhibited a higher growth rate compared to AMPK induction conditions (low glucose). However, cell growth was significantly reduced in conditions with addition of 2DG even in the presence of 25mM glucose (Figure 2A). While SAG treatment increased colony formation in the murine Med1 MB cells, treatment with 2DG ablated colony formation (Figure 2B). Together, these results demonstrate that AMPK regulation of the SHH pathway affects cancer growth in murine MB cells. In addition, reducing GLI1 protein and activity levels through AMPK activation is a promising therapeutic venue for SHH-driven MB treatment.
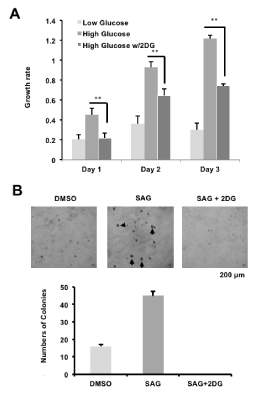
Figure 2. AMPK activation inhibits MB growth (A: Mouse MB Med1 cells were treated with 5 mM (low) glucose, 25 mM (high) glucose or 25 mM glucose with 25 mM 2DG, and subjected to MTT cell growth assays over three days. B: Med1 cells were treated with either DMSO vehicle control, SAG, or SAG and 2DG, and colonies larger than 150um were quantified [*p-value<0.05; **p-value<0.01; ***p-value<0.001])
The SHH pathway is an ubiquitous pathway crucial for cellular proliferation and differentiation, for example, directing both the cellular and morphological development of the cerebellum [1,2]. However, aberrant activation of SHH signaling has been detected in aggressive brain cancers such as glioblastoma multiforme and underlies nearly a third of all MB cases [18,19]. The recent findings of AMPK regulation of GLI1 in human cell lines encourages the study of the AMPK/GLI1 axis in both CNS development as well as in preclinical therapeutic trials. To provide the rationale for these studies in mice, demonstrating the regulation of AMPK on Gli1 in the murine system is necessary.
Several lines of evidence buttress the determination that AMPK regulates mouse Gli1: the finding that AMPK activation in mouse lines suppresses Gli1 expression and/or protein levels in new findings presented here as well as in Li YH, et al. [14] and Di Magno L et al. [15], and the finding that loss of AMPK kinase LKB1 results in elevation of SHH signaling [13]. In addition, the suppression of Gli1 appears to be AMPK-dependent, as knockout of AMPK does not depress Gli1 protein. Lastly, a mechanistic basis for the AMPK/ Gli1 regulation in mice is available: we have both previously shown that β-TrCP-directed proteasome degradation occurs in both mice and humans and that at two phosphorylation sites are candidates for AMPK phosphorylation on mouse Gli1 [15,18].
Disclosure of potential conflicts of interest. None.
Funding. This work was supported by a Showalter Research Scholar grant (207655 to J. -Y. Y.); P30 CA023168 to the Purdue University Center for Cancer Research in support of the use of facilities; Elsa U Pardee Research award and Purdue Start-up Fund (J. -Y.Y.).
- Lee EY, Ji H, Ouyang Z, Zhou B, Ma W, et al. (2010) Hedgehog pathway-regulated gene networks in cerebellum development and tumorigenesis. Proc Natl Acad Sci USA 107: 9736-9741. [Crossref]
- White JJ, Sillitoe 2021 Copyright OAT. All rights reservum: from gene expression patterns to circuit maps. Wiley Interdiscip Rev Dev Biol 2: 149-164. [Crossref]
- Tickle C, Summerbell D, Wolpert L (1975) Positional signalling and specification of digits in chick limb morphogenesis. Nature 254: 199-202. [Crossref]
- Spassky N, Han YG, Aguilar A, Strehl L, Besse L, et al. (2008) Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev Biol 317: 246-259. [Crossref]
- Orentas DM, Hayes JE, Dyer KL, Miller RH (1999) Sonic hedgehog signaling is required during the appearance of spinal cord oligodendrocyte precursors. Development 126: 2419-2429. [Crossref]
- Nery S, Wichterle H, Fishell G (2001) Sonic hedgehog contributes to oligodendrocyte specification in the mammalian forebrain. Development 128: 527-540. [Crossref]
- Dahmane N, Ruiz i Altaba A (1999) Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 126: 3089-3100. [Crossref]
- Corrales JD, Rocco GL, Blaess S, Guo Q, Joyner AL (2004) Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development 22: 5581-5590. [Crossref]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP (1997) Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277: 1109-1113. [Crossref]
- Lee Y, Kawagoe R, Sasai K, Li Y, Russell HR, et al. (2007) Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 26: 6442-6447. [Crossref]
- Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, et al. (2004) The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res 64: 7794-7800. [Crossref]
- Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, et al. (2008) The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res 68: 1768-1776. [Crossref]
- Men Y, Zhang A, Li H, Jin Y, Sun X, et al. (2015) LKB1 Regulates Cerebellar Development by Controlling Sonic Hedgehog-mediated Granule Cell Precursor Proliferation and Granule Cell Migration. Sci Rep 5: 16232. [Crossref]
- Li YH, Luo J, Mosley YY, Hedrick VE, Paul LN, et al. (2015) AMP-Activated Protein Kinase Directly Phosphorylates and Destabilizes Hedgehog Pathway Transcription Factor GLI1 in Medulloblastoma. Cell Rep 12: 599-609. [Crossref]
- Di Magno L, Basile A, Coni S, Manni S, Sdruscia G, et al. (2016) The energy sensor AMPK regulates Hedgehog signaling in human cells through a unique Gli1 metabolic checkpoint. Oncotarget 7: 9538-9549. [Crossref]
- Zhang BB, Zhou G, Li C (2009) AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab 9: 407-416. [Crossref]
- Zhang R, Huang SY, Ka-Wai Li K, Li YH, Hsu WH, et al. (2017) Dual degradation signals destruct GLI1: AMPK inhibits GLI1 through ß-TrCP-mediated proteasome degradation. Oncotarget 8: 49869-49881. [Crossref]
- Braun S, Oppermann H, Mueller A, Renner C, Hovhannisyan A, et al. (2012) Hedgehog signaling in glioblastoma multiforme. Cancer Biol Ther 13: 487-495. [Crossref]
- Northcott PA, Dubuc AM, Pfister S, Taylor MD (2012) Molecular subgroups of medulloblastoma. Expert Rev Neurother 12: 871-884. [Crossref]