The ARF and INK4a genes are located in the same CDKN2a locus, both showing its tumor suppressive activity. ARF has been shown to detect potentially harmful oncogenic signals, making incipient cancer cells undergo senescence or apoptosis. INK4a, on the other hand, responds to signals from aging in a variety of tissues including islets of Langerhans, neuronal cells, and cancer stem cells in general. It also detects oncogenic signals from incipient cancer cells to induce them senescent to prevent neoplastic transformation. Both of these genes are inactivated by gene deletion, promoter methylation, frame shift, and aberrant splicing although mutations that change the coding region affect only the latter. Recent studies indicated that Polycomb genes EZH2 and BMI1 repressed p16INK4a expression in primary cells, but not in cells deficient for pRB protein function. It was also reported that that p14ARF inhibits the stability of the p16INK4a protein in human cancer cell lines and mouse embryonic fibroblasts through its interaction with regenerating islet-derived protein 3γ. Overexpression of INK4a is associated with better prognosis of cancer when it is associated with human papilloma virus infection. However, it has a worse prognostic value in other tumors since it is an indicator of pRB loss. The p16INK4a tumor suppressive protein can thus be used as a biomarker to detect early stage cancer cells as well as advanced tumor cells with pRB inactivation since it is not expressed in normal cells.
p16INK4a, RB, CDKN2a, ARF, PRC, DMTF1, expression, cancer, prognosis, biomarker
Since the discovery as a product of the alternate reading frame of the mouse Arf/Ink4a locus signals, the Arf tumor suppressor has been identified as a key sensor of hyperproliferative stimuli such as those originating from oncoproteins to prevent early stage cancer cells undergo neoplastic transformation by inducing senescence or apoptosis [1,2]. p19Arf and p16Ink4a are transcribed from separate and unique first exons 1β and 1α which splice into two shared exons 2 and 3 (Figure 1). These two genes are different tumor suppressors since p19Arf uses only exons 1 and 2 while p16Ink4a uses all of the exons 1-3 for production of the protein [3,4]. This locus has a very unique genomic structure not found in other mammalian genes due to the unprecedented splicing utilized by Arf which causes an alternate reading frame in the coding region of exon two. Of note this ARF-INK4a (CDKN2a) locus is located 11.5 kbp apart from the genomic locus for CDKN2b that encodes for p15INK4b in humans (Figure 1). The transcriptional regulation for the ARF-INK4a locus has been described [5,6]. The aberrant transcripts from CDKN2a locus have also been reviewed [7]. Both p14ARF and p16INK4a function as tumor suppressors [8-11] despite the lack of amino acid sequence similarity. Consistent with the findings in mice, frequent mutation, promoter methylation, or deletion of the ARF/INK4a locus in human cancers has been reported [5,12,13] second only to p53 in frequency.
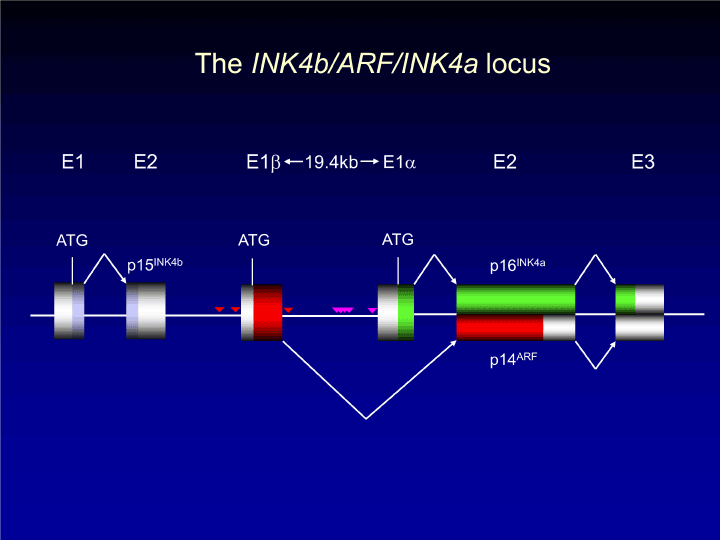
Figure 1. The structure for the human p15INK4b-p14ARF-p16INK4a locus
The genomic structure is well-conserved between human and mice, and thus gene knockout studies have been extensively conducted in mice. The distance between exon 1β and exon 1α is 19.4 kbp in humans and 12.4 kbp in mice. The exon 1α is 3.8 kbp upstream of exon 2 in humans; 5.2 kbp in mice. The ARF-INK4a (CDKN2a) locus is located 11.5 kbp apart from the genomic locus for CDKN2b that encodes for p15INK4b in humans. All of p15Ink4b, p19Arf, and p16Ink4a genes act as tumor suppressors as reported by Krimpenfort, et al. [10, 28]. The reverse triangles indicate the position of Dmp1-binding sites (mouse), which is different on the Arf genomic locus (red: -1060, -183, and +290 from 5’ end of Arf cDNA) from that on Ink4a (pink: -3482, -3390, -3292, -3171, -162 from 5’ end of Ink4a cDNA). The DMP1 consensus is located -2.3 kb and -0.31 kb of ARF and -4.04 kb and -1.40 kb of INK4a in humans. Both of these are Dmp1 target genes [7, 96-98] although the mode of regulation is different [36].
ARF is a highly basic (the predicted pI is 11), insoluble protein which exhibits little structure apart from a pair of α helices at its amino terminus [14]. Ectopic Arf is capable of arresting immortal mouse cell lines such as NIH 3T3 as well as transformed human cells [3,4,15], a classic and requisite property of tumor suppressors. Arf sequesters MDM2 in the nucleolus, preventing p53 degradation [1,2]. Additionally, it inhibits transcription factor E2F activity. These actions lead to cell cycle arrest at G1 and G2 [4]. Importantly, Arf has both p53-dependent and independent functions [1,2,16,17].
Expression of p16INK4a functions to limit cell-cycle progression and to promote cellular senescence in response to multiple stressors, including oncogene activation, telomere erosion, reactive oxygen species, and stalled replication forks [18-22] (Figure 1). Expression of p16INK4a in healthy cells is low, but once induced, p16INK4a binds and inhibits cyclin-dependent kinase 4/6 (CDK4/6) activity, thereby promoting a retinoblastoma (RB) - dependent cell-cycle arrest. This tumor suppressive mechanism is believed to limit the growth of early stage neoplasms, and accordingly, the p16INK4a- CDK4/6-RB signaling is disrupted in most, if not all, human cancers, with inactivation of p16INK4a being the most common lesion of this pathway [12] (Table 1). Although induction of p16INK4a in response to oncogenic stimuli results in a beneficial, anti-cancer mechanism, expression of this tumor suppressor also accelerates mammalian cell aging [19-22]. Both senescent cells and levels of p16INK4a progressively accumulate with age [23,24] and are associated with a decline in the replicative capacity of many tissue types [25,26]. p16Ink4a overexpression has been reported in human cancer and senescent fibroblasts in response to oxidative stress, DNA damage, and changes in chromatin structure [27]. Although gene knock-out mice for p15Ink4b does not show striking phenotypes, it acts as a tumor suppressor when the Arf-Ink4a locus is simultaneously inactivated [28] (Figure 1). Hybrid proteins that encode for p15Ink4b and p16Ink4a have been reported, indicating the synergy of co-deletion for tumor suppressor genes [29].
Table 1. Mechanisms of gene inactivation of the ARF-INK4a locus. The p16INK4a gene/protein could be either underexpressed or overexpressed in human cancer, which will cause inactivation. p16INK4a upregulation detects early stage cancer cells, and thus is associated with better prognosis; however, it can also reflect the RB inactivation in tumors, and is associated with worse prognosis.
Gene |
Mechanism of
inactivation |
Human cancers affected |
Impact on prognosis |
References |
INK4a |
gene deletion |
|
|
5, 12, 13, 18-22 |
|
point mutation |
25-70% of all human cancers |
|
|
|
promoter hypermethylation |
head and neck |
|
|
|
frameshift |
esophagus |
|
|
|
splicing errors |
biliary tract |
|
|
|
|
liver |
|
|
|
|
lung (LOH 36%, methylation 53%) |
|
38 |
|
|
bladder |
|
|
|
|
breast (LOH 9%) |
LOH does not have impact |
33, 34, 88 |
|
|
leukemia |
|
|
|
|
lymphoma |
|
|
|
|
glioblastoma |
|
|
|
|
pancreatic carcinoma (98%) |
|
|
|
|
malignant melanoma |
|
|
INK4a |
overexpression |
HPV (+) cervical, head & neck tumors |
better prognosis |
79-83 |
|
|
colon |
worse prognosis |
32; 86; 92 |
|
|
breast, 20% |
worse prognosis |
33, 34, 88, 93-95 |
|
|
gall bladder |
|
85 |
|
Inverse relationship with RB loss |
breast, lung, endometrium |
possibly worse prognosis |
86-91 |
This review is focused on the mechanism of regulation of INK4a, and its expression in human cancers. Special interests have been put on its value for early stage tumor detection for a potential biomarker since it is not expressed in normal tissues.
Transcriptional regulation plays a major role in p16INK4a regulation as the half-life is long (8 hrs) [30]. A progressive increase of p16INK4a expression has been described in passaged MEFs [8,31], transformation from normal tissue to pre-neoplastic lesions, and from pre-neoplastic lesions to carcinomas [32-34]. Human cells lacking functional pRB contain high levels of p16INK4a mRNA and protein, suggesting a negative feedback loop by which pRB regulates p16INK4a expression in late G1. Hara et al. conducted nuclear run-on assays and promoter characterization of p16INK4a in human fibroblasts [35]. They showed that p16INK4a transcription was affected by the status of RB and defined the p16INK4a promoter that was required for this response. p16INK4a RNA was extremely stable, and the levels did not change during the cell cycle consistent with that fact that it lacked E2F or p53 consensus sequences [35]. Primary human fibroblasts expressed very low levels of p16INK4a, but the mRNA and protein accumulated in late passage, senescent cells [35]. The overexpression of p16INK4a in RB-negative cells is caused by 1) loss of repression by RB, and 2) an increase in the number of population doublings. The mouse p16Ink4a promoter has the Dmp1-binding site in its proximal region, which can be transactivated by Dmp1 in response to cyclin D1 overexpression since cyclin D1 does not have the transactivation domain [36]. The mouse p16Ink4a promoter is repressed by E2F1-3, indicating that p16Ink4a is not overexpressed in RB-deficient cells due to E2F activation. This is consistent with that fact that the Dmp1 upregulates both p19Arf and p16Ink4a, and the p16Ink4a promoter is repressed by E2Fs [37-41] (Inoue, et al. unpublished data).
Ohtani, et al. demonstrated a role for the Ets1 and Ets2 transcription factors based on their ability to activate the p16INK4a promoter through the consensus sequences, and their patterns of expression in human diploid fibroblasts [42]. The induction of p16INK4a by Ets2, which was abundant in young human diploid fibroblasts, was potentiated by signaling through the Ras-Raf-MEK kinase cascade, and inhibited by a direct interaction with the helix-loop-helix protein Id1 [42]. In senescent cells, where the ETS2 levels and MEK signaling decline, they saw a marked increase in p16INK4a expression consistent with the reciprocal reduction of ID1 and accumulation of ETS1 [42]. These results indicated the opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence.
Type 1 diabetes is associated with loss of functional pancreatic β-cells, and restoration of β-cells is a major goal for regenerative therapies [43]. The regenerative capacity of β-cells declines rapidly with age due to accumulation of p16INK4a, resulting in limited capacity for adult endocrine pancreas regeneration [24]. Dhawan, et al. showed that TGFβ signaling via Smad3 integrated with the trithorax complex to activate and maintain Ink4a expression to prevent β-cell replication [43]. Importantly, inhibition of TGFβ signaling resulted in repression of the ARF/INK4a locus, resulting in increased β-cell replication in adult mice. These data revealed a novel role for TGF-β signaling in the regulation of the ARF/INK4a locus. In mice homozygous for a hypomorphic allele of the α-klotho ageing-suppressor gene, accelerated ageing phenotypes were rescued by p16Ink4a deletion, suggesting its dependency [44]. Indeed, p16Ink4a repressed α-klotho promoter activity by blocking the functions of E2Fs, indicating that p16Ink4a plays a role in downregulating α-klotho expression during ageing [44].
Both the expression of p16INK4a and p14ARF are regulated by promoter hypermethylation through proteins of the polycomb repressor complex (PRC1) and PRC2 complexes [45,46] (Figure 2). The polycomb group (PcG) of transcriptional repressor proteins was originally characterized in drosophila for maintenance proteins of pluripotency [47]. PRC2 functions as an initiator of transcription repression and PRC1 functions as a repressor maintenance complex. Thus the methylation mediated by PRC2 is a prerequisite for the binding of PRC1 to the chromatin [45,46,48]. The PRC1 complex is comprised of Polycomb (CXB2, 4, 6-8), Bmi1, HPH (HPH1-3), and RING (RING1 and 2) proteins. The primary mechanism of transcriptional repression by the PRC1 involves mono-ubiquitination of histone H2A K119 by histone H2A ubiquitin ligase [48] (Figure 2). The PRC2 complex is composed of three core proteins, EZH2 which mediates tri-methylation of histone H3K27, EED, SUZ12, and RbAp46 [49] (Figure 2). Histone methyltransferase EZH2 plays a critical role in epigenetic regulation as a bridge between histone methylation/deacetylation and DNA methylation. EZH2 is frequently overexpressed and considered to be an oncogene in cancers [49]. The SET domain of EZH2 possesses methyltransferase activity specific for histone H3K27 [50,51]. The role of deregulated PRC2 in tumor suppressor gene expression, DNA damage response, and the fidelity of DNA replication has been suggested [52], resulting in long-term reversible suppression p14ARF and p16INK4a.
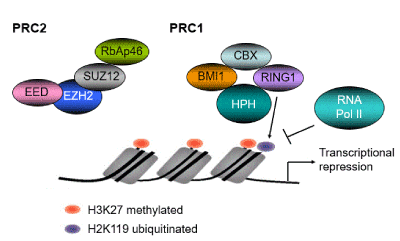
Figure 2. PRC1 and PRC2 repress transcription from the ARF-INK4a locus
Both the expression of p14ARF and p16INK4a are regulated by promoter hypermethylation through proteins of the polycomb repressor complex (PRC1) and PRC2 complexes [45-48]. The polycomb group of transcriptional repressor proteins was originally characterized in drosophila for maintenance proteins of pluripotency [47]. PRC1 functions as a repressor of the maintenance complex while PRC2 functions as an initiator of transcriptional repression. Thus the methylation mediated by PRC2 is a prerequisite for the binding of PRC1 to the chromatin [48]. The PRC1 complex is comprised of Polycomb (CXB2, 4, 6-8), Bmi1, HPH (HPH1-3), and RING (RING1 and 2) proteins. The most important mechanism for transcriptional repression by the PRC1 complex involves mono-ubiquitination of histone H2A K119 by histone H2A ubiquitin ligase (48). The PRC2 complex is composed of three core proteins, EZH2 which mediates tri-methylation of histone H3K27, EED, SUZ12, and RbAp46 [49].
The ARF/INK4a tumor suppressor locus, which is a key executor of cellular senescence, is regulated by members of the PcG of transcriptional repressors (Figure 2). Barradas, et al. [53] showed that signaling from oncogenic RAS overrides PcG-mediated repression of INK4a by activating the H3K27 demethylase JMJD3, and down-regulating the methyltransferase EZH2. In human fibroblasts, JMJD3 activated p16INK4a, but not p14ARF, and caused p16INK4a-dependent cell cycle arrest. In MEFs, Jmjd3 activated both Ink4a and Arf and causes a p53-dependent arrest, echoing the effects of Ras in this system. Their findings directly implicate JMJD3 in the regulation of ARF/INK4a during oncogene-induced senescence (OIS), suggesting that JMJD3 has the capacity to act as a tumor suppressor [53]. Similar findings were also reported from two other groups, indicating the importance of EZH2 - mediated repression of the ARF/INK4a locus in cellular senescence [54,55].
The p16Ink4a protein is always upregulated in Rb-deficient cells [35,56]. So, what are the molecular mechanisms? It was believed that dysregulated activities of E2F proteins were responsible for overexpression of p16Ink4a in Rb-deficient cells, but it is the p19Arf promoter that is transactivated with E2F1-3 overexpression, not p16Ink4a. Indeed E2F1-3 repressed the murine p16Ink4a promoter and E2F4, even combined with nuclear localization signal, had no effect (Inoue, et al. unpublished data). This indicates that it is not E2Fs that is responsible for overexpression of p16Ink4a in Rb-deficient cells.
Genetic studies have demonstrated that Bmi1 promotes cell proliferation and stem cell self-renewal with a correlative decrease of p16Ink4a expression. Kotake, et al. demonstrate that Polycomb genes EZH2 and BMI1 repressed p16INK4a expression in human and mouse primary cells, but not in cells deficient for pRB protein function [57]. The p16INK4a locus was H3K27-methylated and bound by BMI1, RING2, and SUZ12 [48]. Inactivation of pRB family proteins abolished H3K27 methylation and disrupted BMI1, RING2, and SUZ12 binding to the INK4a-ARF locus, associated with a substantial increase of p16INK4a. qRT-PCR analysis showed that among the four INK4 and ARF genes, only p16INK4a mRNA was significantly increased in WI38 cells when BMI1 was silenced [57]. The level of p18INK4c and p14ARF mRNA was slightly decreased by BMI1 silencing. BMI1 knockdown resulted in slower cell growth associated with p16INK4a increase. Together, these results demonstrate that p16INK4a seems to be a specific target of BMI1 function in human primary cells, confirming the difference between mouse and human cells for the relative importance for p19Arf and p16Ink4a [8-13]. Their results raised a model in which pRB recruit PRC2 to trimethylate p16INK4a, priming the BMI1-containing PRC1L ubiquitin ligase complex to silence p16INK4a, explaining why p16INK4a levels are high in RB-deficient cells [57].
Kobayashi, et al. [30] found that that p14ARF regulates the stability of the p16INK4a protein in human cancer cell lines as well as in MEFs. In particular, ARF promoted rapid degradation of p16INK4a protein (the half-life became 3.5 hrs from 8 hrs), which was mediated by the proteasome and, more specifically, by interaction of ARF with one of its subunits, regenerating islet-derived protein 3γ (REGγ). Furthermore, this ARF-dependent destabilization of p16INK4a was abrogated by knock-down of REGγ or by pharmacological blockade of its nuclear export. Thus, their findings uncovered a novel crosstalk of two key tumor suppressors mediated by a REGγ-dependent mechanism [30].
Several pieces of evidence suggested that the ability to bypass senescence is the main molecular mechanism in the progression of pre-malignant to fully malignant cells [58,59]. This hypothesis is based on the concept of OIS, which was established after demonstration of p53- and p16Ink4a-mediated senescent-like growth arrest in response to expression of oncogenic Ras in normal primary cells [18,58-60], which has been considered as highly possible mechanism to prevent proliferation of incipient cancer cells. Consistently, senescent cells have been reported in a number of different benign lesions, including nevi and neurofibromas [61,62], but not in cancer. Since p16Ink4a is involved in OIS, the overexpression has been found in benign and pre-malignant lesions with senescent cells where the ARF/INK4a locus plays its role [61-63]. Indeed, human nevus lesions remain in a growth-arrested state, and rarely progress to malignant melanomas [64,65]. A mutation in the downstream effector of Ras, BRAF (BRAFV600E) is often found in malignant melanomas, the expression of which in human melanocytes induces cell cycle arrest, followed by p16INK4a induction [61-65]. A similar finding has been reported in schwannomas/neurofibromas [64]. Schwannomas express high levels of p16INK4a, and show senescence-associated β-galactosidase activity, BRAF mutations, and very low proliferative activity [66]. Conversely, the malignant counterparts for these tumors are negative for p16INK4a, and loss of p16INK4a has thus been implicated in the development of both types of malignant neoplasms [64-68]. In other words, benign tumors overexpress p16INK4a, which seems to inhibit cell proliferation in response to oncogenic stimuli, protecting cells from malignant transformation. p16INK4a methylation could thus have a diagnostic value; it can be used in the differential diagnosis from pre-malignant and malignant lesions [69]. In fact, p16INK4a methylation is found of 53.2 % of non-small cell lung cancer while p14ARF methylation is found 6.5 % [38], the frequency 8 times higher than that in ARF.
OIS is emerging as a potent cancer-protective response to oncogenic events, serving to eliminate early neoplastic cells from the neoplastic tissue. Kuilman, et al. [70] reported a unique role of interleukin-6 (IL-6) in OIS of cancer cells. They found that OIS was linked specifically to the activation of an inflammatory transcriptome. Induced genes included IL-6, which upon secretion by senescent cells acted mitogenically in a paracrine fashion [70]. IL-6 was also required for the execution of OIS, but in a cell-autonomous fashion. Its depletion caused the inflammatory network to collapse and abolished senescence entry and maintenance. They also demonstrated that C/EBPβ cooperated with IL-6 to amplify the activation of the inflammatory network, including IL-8. In human colon adenomas, IL-8 specifically colocalized with p16INK4a-positive epithelium [70]. They proposed a model in which interleukins connect senescence with an inflammatory phenotype and cancer. The role of IL-6 in OIS has been confirmed by later studies [71,72], indicating the unique role of this cytokine in cancer prevention.
Concerning the role of p16INK4a in OIS, Burd, et al. has made a very intriguing report [73]. They have made a luciferase knock-in mouse (p16LUC), which faithfully reports expression of p16INK4a. Lifelong assessment of luminescence in p16+/LUC mice revealed an exponential increase with aging, which was highly variable in a cohort of contemporaneously housed, syngeneic mice. However, the expression of p16INK4a with aging did not predict cancer development, suggesting that the accumulation of senescent cells is not a principal determinant of cancer-related death. Importantly, in 14 of 14 (e.g. C3(1)TAg cells, mammary tumor: MMTV-HER2/neu, K14-CRE; p53Lox/Lox, pancreatic cancer: Pdx-CRE; LSL-KrasG12D; p53Lox/Lox, B cell lymphoma: Eµ-Myc), and endometrial cancer (Sprr2f-CRE; Lkb1Lox/Lox) tested tumor models, the expression of p16LUC was focally activated by early neoplastic events, enabling visualization of tumors with sensitivity exceeding other imaging modalities [73]. Activation of p16Ink4a was noted in the emerging neoplasm and surrounding stromal cells. They concluded that p16Ink4a activation is a characteristic of all emerging cancers, making the p16LUC allele a sensitive, unbiased reporter of neoplastic transformation. It seems p16Ink4a is a gateway to detect oncogenic signals to force incipient cancer cells into senescence and/or apoptosis from this study; however, Zindy, et al. had reported that p19Arf was the major player that played its role in vivo and in vitro in response to c-Myc overexpression since p16Ink4a was not induced in response to c-Myc [74,75]. Our data indicate that the p16Ink4a promoter is repressed in response to E2F1-3 although the p19Arf promoter was strongly activated by these E2Fs [6,76]. Moreover, our HER2 study showed that p16Ink4a was induced 2-16 folds (median 3 folds) while p19Arf was induced 7-39 folds (median 12 folds) in wild type Dmp1 ND tumors [40] indicating that p19Arf induction was the predominant player for quenching the oncogenic stress in tumors driven by c-Myc, E2Fs, and HER2 in the murine system.
It should be noted that the ARF protein is always overexpressed whenever the mRNA is high. However, that is not the case in p16INK4a. Kobayashi, et al. reported that the p16INK4a protein is unstable due to the increased turnover in ARF overexpressing cells, suggesting the crosstalk between ARF and p16INK4a, and suggested possible discrepancy between p16INK4a mRNA and protein [30]. Hence, analysis of p16INK4a protein levels is necessary to establish its role in tumor suppression in those overexpressing ARF. The impact of aging should also be considered in the assessment of p16Ink4a in aged mice [19-22]. As a matter of fact, p16Ink4a remains high in MEFs that are passaged for more than 12 days (passage 4) regardless of the protocol [8,31]; it remains consistently elevated in later passages regardless of the mechanism of immortalization indicating the Cdk4 inhibition is not enough to inhibit cell proliferation in immortalized MEFs. Whether ARF or INK4a is major player in silencing oncogenic signals remains a big question that should be resolved in human systems.
The RB protein is inactivated by interaction with the high-risk HPV oncoprotein E7 [77,78], and oncoprotein E6 induces degradation of the tumor suppressor p53. RB inactivation releases p16INK4a from its negative feedback control, causing a paradoxical increase in the levels of this protein, which attempts to inhibit uncontrolled cellular replication. As a consequence, p16INK4a is overexpressed in HPV-expressing tumors such as cervical cancer and head and neck tumors [79-81] (Table 1).
Previous reports suggested that p16INK4a immunostaining allows precise identification of even small cervical intraepithelial neoplasia or cervical cancer lesions in biopsies. The prognostic value of overexpressed p16INK4a in cervical cancer has been evaluated for several years with controversial results. Lin, et al. performed meta-analyses of studies assessing the clinical and prognostic significance of overexpression of p16INK4a in cervical cancer to evaluate the prognostic value of overexpressed p16INK4a in cervical cancer (15 publications; 1,633 cases) [79]. Analysis showed that p16INK4a overexpression was not significantly associated with tumor TNM staging, tumor grade, tumor size, or vascular invasion. However, overexpression of p16INK4a was highly correlated with no lymph node metastasis, increased overall survival (p = 0.002) and increased disease-free survival (p = 0.001). They showed that overexpression of p16INK4a in cervical cancer was connected with better prognosis of cervical cancer: increased overall and disease-free survival, consistent with previously published studies [80]. Coordes, et al. conducted a meta-analysis (18 articles, 4,424 patients) between HPV/p16 and clinical outcomes in head and neck squamous cell carcinoma (HNSCC; ref. 81). The meta-analysis showed a significantly improved 5-year overall survival (OS), 5-year disease-free survival and their corresponding hazard ratio for HPV(+)/p16(+) HNSCC in comparison to other groups. It is generally accepted that p16INK4a expression is frequently associated with HPV infection and a better prognostic factor in HNSCC [82,83] since they respond to chemo/radiotherapy.
Aberrant expression of p16INK4a, either high or low, is found frequently in human cancers. In colon cancer, a very low p16INK4a immunostaining in normal mucosa with a progressively higher expression in aberrant crypt foci, non-serrated adenomas, further increase in primary carcinomas and metastatic tumors have been reported [32] (Table 1). A similar pattern was reported in skin cancer where p16INK4a expression increases from relatively low levels in pre-malignant lesions to high levels in in situ and invasive squamous cell carcinomas [84]. In breast cancer, p16INK4a was negative or low in normal ductal epithelium, but a progressive increase was reported in benign lesions and carcinoma [33,34]. Increased nuclear p16INK4a protein expression compared with normal epithelium has also been reported in pre-neoplastic and tumor tissues of the gallbladder [85]. Therefore, p16INK4a induction as a protective mechanism for tumor development must be present in humans in relation to senescence, and inactivation of the p16INK4a - RB pathway should be present in cases where a progressive increase of p16INK4a is seen from pre-malignant lesions (RB wild type) to malignant neoplasia (RB loss). In this sense, inactivation of RB results in increased p16INK4a expression in tumor cells due to corrupted feedback loop [86] as reported in an early study. Indeed, RB-loss is a frequent event in many neoplasms and it is associated with uncontrolled cell proliferation. Loss of heterozygosity (LOH) of RB was found in 39 % of breast cancers [87]. LOH of the ARF/INK4a locus was found in 20 % of breast cancer without association of the prognosis [88]. In the former samples, high p16INK4a protein expression was observed with RB LOH cases. An inverse relationship between p16INK4a and RB expression has been reported in breast cancer [87], lung cancer [89,90], and endometrial cancers [91] (Table 1). In short, the negative feed-back loop between RB and p16INK4a could explain overexpression of the p16INK4a in malignant tumors showing uncontrolled cell proliferation. The molecular mechanisms that explain p16INK4a overexpression in RB-deficient cells are explained before this section.
In colon cancer, p16INK4a overexpression was associated with clinical features of a worse prognosis, such as sex, distal location, tumor grade and stage [92] (Table 1). In breast cancer, p16INK4a overexpression was detected in about 20 % of tumors and was significantly associated with unfavorable prognostic indicators, such as high grade and negative estrogen receptor status [93]. Garcia et al. showed that simultaneous overexpression of p73 and p16INK4a was correlated with the lymph node metastasis, positivity for p53, vascular invasion, and negative progesterone receptors [94]. Kerlikowske, et al. described the association between p16INK4a overexpression in breast ductal carcinoma in situ and the risk of subsequent DCIS or invasive cancer [95]. In short, p16INK4a overexpression, together with a high Ki67 and COX-2 expression, was associated with progression to an invasive breast cancer, whereas p16INK4a overexpression with high Ki67 but without COX-2 expression was associated with DCIS. These results raise the idea that more molecular events than p16INK4a overexpression are needed in malignant transformation of tumors, and that p16INK4a overexpression is generally a bad prognostic marker in HPV (-) cells because it leads to RB inactivation.
We have reviewed p16INK4 alterations in cancer. Both ARF and INK4a genes are inactivated in human cancer by gene deletion, promoter methylation, gene frame shift, and splicing errors although mutations mainly affect only the latter [12,13]. It has also been reported that many human cancers overexpress p16INK4a [84-86,92-94]. The mechanism of p16INK4a overexpression in RB-null cells is related to aberrant PRC activity [57] rather than hyperstimulated E2Fs. Tumors that overexpress p16INK4a should have better prognosis than the others in early stage since they stop cell proliferation due to inhibition of the cell cycle by activating RB. Conversely, if p16INK4a is overexpressed in tumor cells as a result of RB-loss, such patients should have worse prognosis as shown in published studies. The exception for this rule is overexpression of p16INK4a in human cervical and neck cancers with HPV; the prognosis of such patients is better than the others since they respond to therapies. In summary, we have to be cautious in using p16INK4a as a biomarker for cancer, since it can be associated with either favorable (early stage or HPV - positive tumors) or worse prognosis (HPV - negative tumors).
We thank all other members of Dr. Inoue’s lab for sharing unpublished research data.
K. Inoue was supported by NIH/NCI 2R01CA106314, ACS RSG-07-207-01-MGO, and KG080179.
The authors declare no conflicts of interest.
- Maggi LB Jr, Winkeler CL, Miceli AP, Apicelli AJ, Brady SN, et al. (2014) ARF tumor suppression in the nucleolus. Biochim Biophys Acta 1842: 831-839. [Crossref]
- Basu S, Murphy ME (2016) Genetic Modifiers of the p53 Pathway. Cold Spring Harb Perspect Med 6: a026302. [Crossref]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ (1995) Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 83: 993-1000. [Crossref]
- Quelle DE, Cheng M, Ashmun RA, Sherr CJ (1997) Cancer-associated mutations at the INK4a locus cancel cell cycle arrest by p16INK4a but not by the alternative reading frame protein p19ARF. Proc Natl Acad Sci USA 94: 669-673. [Crossref]
- Gil J, Peters G (2006) Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol 7: 667-677. [Crossref]
- Inoue K, Roussel MF, Sherr CJ (1999) Induction of ARF tumor suppressor gene expression and cell cycle arrest by transcription factor DMP1. Proc Natl Acad Sci USA 96: 3993-3998. [Crossref]
- Inoue K, Fry EA (2016) Aberrant splicing of the DMP1-ARF-MDM2-p53 pathway in cancer. Int J Cancer 139: 33-41. [Crossref]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, et al. (1997) Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 91: 649-659. [Crossref]
- Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ (1999) Tumor spectrum in ARF-deficient mice. Cancer Res 59: 2217-2222. [Crossref]
- Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A (2001) Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature 413: 83-86. [Crossref]
- Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, et al. (2001) Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 413: 86-91. [Crossref]
- Ruas M, Peters G (1998) The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta 1378: F115-F177.
- Sharpless NE, Bardeesy N, Lee KH (1997) Cancer-associated mutations at the INK4a locus cancel cell cycle arrest by p16INK4a but not by the alternative reading frame protein p19ARF. Proc Natl Acad Sci USA 94: 669-673.
- Kuo ML, den Besten W, Bertwistle D, Roussel MF, Sherr CJ (2004) N-terminal polyubiquitination and degradation of the Arf tumor suppressor. Genes Dev 18: 1862-1874. [Crossref]
- Stott FJ, Bates S, James MC, McConnell BB, Starborg M, et al. (1998) The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J 17: 5001-5014.
- Weber JD, Jeffers JR, Rehg JE, Randle DH, Lozano G, et al. (2000) p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev 14: 2358-2365. [Crossref]
- Sherr CJ, Bertwistle D, DEN Besten W, Kuo ML, Sugimoto M, et al. (2005) p53-Dependent and -independent functions of the Arf tumor suppressor. Cold Spring Harb Symp Quant Biol 70: 129-137. [Crossref]
- McMahon M, Woods D (2001) Regulation of the p53 pathway by Ras, the plot thickens. Biochim Biophys Acta 1471: M63-M71.
- Kim WY, Sharpless NE (2006) The regulation of INK4/ARF in cancer and aging. Cell 127: 265-275. [Crossref]
- Sharpless NE, Sherr CJ (2015) Forging a signature of in vivo senescence. Nature Rev Cancer 15: 397-408.
- Sherr CJ, Beach D, Shapiro GI (2016) Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov 6: 353-367.
- He S, Sharpless NE (2017) Senescence in Health and Disease. Cell 169: 1000-1011. [Crossref]
- Zindy F, Quelle DE, Roussel MF, Sherr CJ (1997) Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 15: 203-211. [Crossref]
- Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, et al. (2006) p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 443: 453-457. [Crossref]
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, et al. (2006) Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 443: 421-426. [Crossref]
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, et al. (2006) Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443: 448-452. [Crossref]
- Romagosa C, Simonetti S, López-Vicente L, Mazo A, Lleonart ME, et al. (2011) p16Ink4a overexpression in cancer: a tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 30: 2087-2097. [Crossref]
- Krimpenfort P, Ijpenberg A, Song JY, van der Valk M, Nawijn M, et al. (2007) p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature 448: 943-946. [Crossref]
- Tian X, Azpurua J, Ke Z, Augereau A, Zhang ZD, et al. (2015) INK4 locus of the tumor-resistant rodent, the naked mole rat, expresses a functional p15/p16 hybrid isoform. Proc Natl Acad Sci USA 112: 1053-1058. [Crossref]
- Kobayashi T, Wang J, Al-Ahmadie H, Abate-Shen C (2013) ARF regulates the stability of p16 protein via REGγ-dependent proteasome degradation. Mol Cancer Res 11: 828-833. [Crossref]
- Inoue K, Wen R, Rehg JE, Adachi M, Cleveland JL, et al. (2000) Disruption of the ARF transcriptional activator DMP1 facilitates cell immortalization, Ras transformation, and tumorigenesis. Genes Dev 14: 1797-809. [Crossref]
- Dai CY, Furth EE, Mick R, Koh J, Takayama T, et al. (2000) p16(INK4a) expression begins early in human colon neoplasia and correlates inversely with markers of cell proliferation. Gastroenterol 119: 929-942. [Crossref]
- Milde-Langosch K, Bamberger AM, Rieck G, Kelp B, Löning T (2001) Overexpression of the p16 cell cycle inhibitor in breast cancer is associated with a more malignant phenotype. Breast Cancer Res Treat 67: 61-70. [Crossref]
- Di Vinci A, Perdelli L, Banelli B, Salvi S, Casciano I, et al. (2005) p16(INK4a) promoter methylation and protein expression in breast fibroadenoma and carcinoma. Int J Cancer 114: 414-421. [Crossref]
- Hara E, Smith R, Parry D, Tahara H, Stone S, et al. (1996) Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol 16: 859-67. [Crossref]
- Zhu S, Mott RT, Fry EA, Taneja P, Kulik G, et al. (2013) Cooperation between cyclin D1 expression and Dmp1-loss in breast cancer. Am J Pathol 183: 1339-1350. [Crossref]
- Sreeramaneni R, Chaudhry A, McMahon M, Sherr CJ, Inoue1 K (2005) Ras-Raf-Arf signaling critically depends on the Dmp1 transcription factor. Mol Cell Biol 25: 220-232.
2021 Copyright OAT. All rights reserv
- Mallakin A, Sugiyama T, Taneja P, Matise LA, Frazier DP, et al. (2007) Mutually exclusive inactivation of DMP1 and ARF/p53 in lung cancer. Cancer Cell 12: 381-394. [Crossref]
- Mallakin A, Sugiyama T, Kai F, Taneja P, Kendig RD, et al. (2010) The Arf-inducing transcription factor Dmp1 encodes transcriptional activator of amphiregulin, thrombospondin-1, JunB and Egr1. Int J Cancer 126: 1403-1416. [Crossref]
- Taneja P, Maglic D, Kai F, Sugiyama T, Kendig RD, et al. (2010) Critical roles of DMP1 in human epidermal growth factor receptor 2/neu-Arf-p53 signaling and breast cancer development. Cancer Res 70: 9084-9094. [Crossref]
- Fry EA, Taneja P, Maglic D, Zhu S, Sui G, et al. (2013) Dmp1α inhibits HER2/neu-induced mammary tumorigenesis. PLoS One 8: e77870. [Crossref]
- Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, et al. (2001) Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 409: 1067-1070. [Crossref]
- Dhawan S, Dirice E, Kulkarni RN, Bhushan A (2016) Inhibition of TGF-β Signaling Promotes Human Pancreatic β-Cell Replication. Diabetes 65: 1208-1218. [Crossref]
- Sato S, Kawamata Y, Takahashi A, Imai Y, Hanyu A, et al. (2015) Ablation of the p16(INK4a) tumour suppressor reverses ageing phenotypes of klotho mice. Nat Commun 6: 7035. [Crossref]
- Blackledge NP, Rose NR, Klose RJ (2015) Targeting Polycomb systems to regulate gene expression: modifications to a complex story. Nature Rev Mol Cell Biol 16: 643-649.
- Piunti A, Shilatifard A (2016) Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 352: aad9780. [Crossref]
- Lanzuolo C, Orlando V (2012) Memories from the polycomb group proteins. Annu Rev Genet 46: 561-589. [Crossref]
- Rayess H, Wang MB, Srivatsan ES (2012) Cellular senescence and tumor suppressor gene p16. Int J Cancer 130: 1715-1725.
- Christofides A, Karantanos T, Bardhan K, Boussiotis VA (2016) Epigenetic regulation of cancer biology and anti-tumor immunity by EZH2. Oncotarget 7: 85624-85640. [Crossref]
- Tiffen J, Gallagher SJ, Hersey P (2015) EZH2: an emerging role in melanoma biology and strategies for targeted therapy. Pigment Cell Melanoma Res 28: 21-30. [Crossref]
- Katoh M (2016) Mutation spectra of histone methyltransferases with canonical SET domains and EZH2-targeted therapy. Epigenomics 8: 285-305.
- Liu X, Wu Q, Li L (2017) Functional and therapeutic significance of EZH2 in urological cancers. Oncotarget 8: 38044-38055. [Crossref]
- Barradas M, Anderton E, Acosta JC, Li S, Banito A, et al. (2009) Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev 23: 1177-1182. [Crossref]
- Tzatsos A, Pfau R, Kampranis SC, Tsichlis PN (2009) Ndy1/KDM2B immortalizes mouse embryonic fibroblasts by repressing the Ink4a/Arf locus. Proc Natl Acad Sci U S A 106: 2641-2646. [Crossref]
- Agherbi H, Gaussmann-Wenger A, Verthuy C, Chasson L, Serrano M, et al. (2009) Polycomb mediated epigenetic silencing and replication timing at the ARF/INK4a locus during senescence. PLoS One 4: e5622. [Crossref]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, et al. (1992) Effects of an Rb mutation in the mouse. Nature 359: 295-300. [Crossref]
- Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, et al. (2007) pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev 21: 49-54. [Crossref]
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, et al. (2005) Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436: 660-665. [Crossref]
- Collado M, Blasco MA, Serrano M (2007) Cellular senescence in cancer and aging. Cell 130: 223-233. [Crossref]
- Sinha VC, Qin L, Li Y (2015) A p53/ARF-dependent anticancer barrier activates senescence and blocks tumorigenesis without impacting apoptosis. Mol Cancer Res 13: 231-8.
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, et al. (2005) BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436: 720-724. [Crossref]
- Courtois-Cox S, Jones SL, Cichowski K (2008) Many roads lead to oncogene-induced senescence. Oncogene 27: 2801-2809. [Crossref]
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, et al. (2004) Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114: 1299-1307. [Crossref]
- Sargen MR, Merrill SL, Chu EY, Nathanson KL (2016) CDKN2A mutations with p14 loss predisposing to multiple nerve sheath tumours, melanoma, dysplastic naevi and internal malignancies: a case series and review of the literature. Br J Dermatol 175: 785-789. [Crossref]
- Joselow A, Lynn D, Terzian T, Box NF (2017) Senescence-Like Phenotypes in Human Nevi. Methods Mol Biol 1534: 175-184. [Crossref]
- Romagosa C, Simonetti S, Serrano C (2009) Senescence markers in Schwannomas. Virchows Archiv 455: 372.
- Perrone F, Tabano S, Colombo F, Dagrada G, Birindelli S, et al. (2003) p15INK4b, p14ARF, and p16INK4a inactivation in sporadic and neurofibromatosis type 1-related malignant peripheral nerve sheath tumors. Clin Cancer Res 9: 4132-4138. [Crossref]
- Damsky W, Micevic G, Meeth K, Muthusamy V, Curley DP, et al. (2015) mTORC1 activation blocks BrafV600E-induced growth arrest but is insufficient for melanoma formation. Cancer Cell 27: 41-56. [Crossref]
- Feng W, Han Z, Zhu R, Liu P, Liu S (2015) Association of p16 gene methylation with prostate cancer risk: a meta-analysis. J BUON 20: 1074-1080. [Crossref]
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, et al. (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133: 1019-1031. [Crossref]
- Atwood AA, Sealy L (2010) Regulation of C/EBPbeta1 by Ras in mammary epithelial cells and the role of C/EBPbeta1 in oncogene-induced senescence. Oncogene 29: 6004-6015.
- Zacarias-Fluck MF, Morancho B, Vicario R, Luque Garcia A, Escorihuela M, et al. (2015) Effect of cellular senescence on the growth of HER2-positive breast cancers. J Natl Cancer Inst 107. [Crossref]
- Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, et al. (2013) Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 152: 340-351. [Crossref]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, et al. (1998) Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev 12: 2424-2433. [Crossref]
- Zindy F, Williams RT, Baudino TA, Rehg JE, Skapek SX, et al. (2003) Arf tumor suppressor promoter monitors latent oncogenic signals in vivo. Proc Natl Acad Sci USA 100: 15930-15935. [Crossref]
- Bates S, Phillips AC, Clark PA, Stott F, Peters G, et al. (1998) p14ARF links the tumour suppressors RB and p53. Nature 395: 124-125. [Crossref]
- Munger K, Werness BA, Dyson N (1989) Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J 8: 4099-105.
- Huang PS, Patrick DR, Edwards G, Goodhart PJ, Huber HE, et al. (1993) Protein domains governing interactions between E2F, the retinoblastoma gene product, and human papillomavirus type 16 E7 protein. Mol Cell Biol 13: 953-60. [Crossref]
- Lin J, Albers AE, Qin J, Kaufmann AM (2014) Prognostic significance of overexpressed p16INK4a in patients with cervical cancer: a meta-analysis. PLoS One 9: e106384. [Crossref]
- Masoudi H, Van Niekerk D, Gilks C (2006) Loss of p16INK4 expression in invasive squamous cell carcinoma of the uterine cervix is an adverse prognostic marker. Histopathol 49: 542-545.
- Coordes A, Lenz K, Qian X, Lenarz M, Kaufmann AM, et al. (2016) Meta-analysis of survival in patients with HNSCC discriminates risk depending on combined HPV and p16 status. Eur Arch Otorhinolaryngol 273: 2157-2169. [Crossref]
- Nevens D, Nuyts S (2015) HPV-positive head and neck tumours, a distinct clinical entity. B-ENT 11: 81-87.
- Sedghizadeh PP, Billington WD, Paxton D, Ebeed R, Mahabady S, et al. (2016) Is p16-positive oropharyngeal squamous cell carcinoma associated with favorable prognosis? A systematic review and meta-analysis. Oral Oncol 54: 15-27. [Crossref]
- Nilsson K, Svensson S, Landberg G (2004) Retinoblastoma protein function and p16INK4a expression in actinic keratosis, squamous cell carcinoma in situ and invasive squamous cell carcinoma of the skin and links between p16INK4a expression and infiltrative behavior. Mod Pathol 17: 1464-1474.
- Kriegl L, Neumann J, Vieth M (2008) Expression of the p16(INK4a) gene product in premalignant and malignant epithelial lesions of the gallbladder. Ann Diagn 12: 161-164.
- Schwartz B, Avivi-Green C, Polak-Charcon S (1998) Sodium butyrate induces retinoblastoma protein dephosphorylation, p16 expression and growth arrest of colon cancer cells. Mol Cell Biochem 188: 21-30.
- Herschkowitz JI, He X, Fan C (2008) The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res 10: R75.
- Maglic D, Zhu S, Fry EA, Taneja P, Kai F, et al. (2013) Prognostic value of the hDMP1-ARF-Hdm2-p53 pathway in breast cancer. Oncogene 32: 4120-4129. [Crossref]
- Leversha MA, Fielding P, Watson S, Gosney JR, Field JK (2003) Expression of p53, pRB, and p16 in lung tumours: a validation study on tissue microarrays. J Pathol 200: 610-619. [Crossref]
- Wikenheiser-Brokamp KA (2006) Retinoblastoma regulatory pathway in lung cancer. Current Mol Med 6: 783-793.
- Koh VM, Shi YX, Tang QH (2011) p16 and retinoblastoma protein expression in endometrial carcinoma and clinical significance. Euro J Gynaecol Oncol 32: 309-315.
- Lam AK, Ong K, Giv MJ, Ho YH (2008) p16 expression in colorectal adenocarcinoma: marker of aggressiveness and morphological types. Pathology 40: 580-585. [Crossref]
- Milde-Langosch K, Bamberger AM, Rieck G, Kelp B, Lning T (2001) Overexpression of the p16 cell cycle inhibitor in breast cancer is associated with a more malignant phenotype. Breast Cancer Res Treat 67: 61-70. [Crossref]
- Garcia V, Silva J, Dominguez G, García JM, Peña C, Rodriguez R, et al. (2004) Overexpression of p16INK4a correlates with high expression of p73 in breast carcinomas. Mutat Res 554: 215-221.
- Kerlikowske K, Molinaro AM, Gauthier ML, Berman HK, Waldman F, et al. (2010) Biomarker expression and risk of subsequent tumors after initial ductal carcinoma in situ diagnosis. J Natl Cancer Inst 102: 627-637. [Crossref]
- Inoue K, Fry EA, Frazier DP (2016) Transcription factors that interact with p53 and Mdm2. Int J Cancer 138: 1577-1585. [Crossref]
- Fry EA, Taneja P, Inoue K (2017) Oncogenic and tumor-suppressive mouse models for breast cancer employing HER2/neu. Int J Cancer 140: 495-503.
- Inoue K, Fry EA (2017) Haploinsufficient tumor suppressor genes. Adv Med Biol 118: 83-122. [Crossref]