Soft tissue and visceral sarcomas are rare tumors which often develop in muscles, joints, nerves, fat, skin, or vessels. In a significant proportion, they appear in the abdomen and in the female reproductive system. Considering that early-stage sarcomas do not give symptoms, there are often late diagnosed in a quite advanced and/or metastatic stage. Malignant peripheral nerve sheath tumors (MPNSTs) are an uncommon type of malignant soft tissue sarcomas. In this report, we refer to a rare case of histologically confirmed MPNST arisen from corpus uterus in a 74 years old woman. The above-mentioned patient was admitted with abdominal pain and severe vaginal bleeding. Firstly, she underwent emergency endometrial ablation and removal of the tumor that was protruding from the endometrial cavity. Immediately after receiving histological diagnosis, abdominal hysterectomy was performed with bilateral oophorectomy. Considering that the available published literature has only a few reports of MPNST mainly arising from the uterine cervix, with no established treatment, our contribution could help the scientific community in adding data to MPNSTs arising from the uterine body.
malignant nerve tumor, uterus tumor, corpus uterine tumor, MPNST
Malignant peripheral nerve sheath tumor (MPNST) is a connective tissue neoplasm which is classified as sarcoma. MPNST with a coexistence of rhabdomyoblastoma component are well known as malignant tertiary tumors [1,2]. MPNST symptoms may include swelling of the limbs (hands or feet), that is also known as peripheral edema, which will be painless, difficulty moving the tumor-bearing end, including the injury, pain located at the tumor site or at the extremity [3,4]. Moreover, some of the neurological symptoms are: Pain or discomfort, numbness, burning or "pins and needles", dizziness and / or loss of balance [3,4].
MPNST make up about 5-10% of soft tissue sarcomas in adults and are rare in children [5]. Furthermore, they may occur as isolated tumors or in neurofibromatosis type 1 (NF1) [5]. About half of the cases are diagnosed in people with neuropsychiatric disorders. The lifetime risk is between 8-13% for an MPNST in patients with type 1 neuroimaging [6]. Their incidence in NF1 patients ranges and is approximately 2-5%. Over 50% of MPNST patients also have NF1 [7-8]. They are often extend locally and located on the trunk and extremities. Patients with a slight loss of neurofibromine are more likely to have cutaneous neuromas at younger ages and are potentially associated with a higher incidence of MPNST than the general population of NF1 patients. A variety of different genetic alterations are reported in the cases of MPNST, but it is not clear whether any of them are causally related to oncogenesis or malignancy. It is thought that loss of p53 activity contributes to the molecular pathogenesis of MPNSTs, as p53 reacts immunohistochemically in more than half of patients [7]. NF1 coexists in more than half of MPNST patients. Patients with NF1 are at greater risk of developing sarcoma 10-20 years after the onset of neurinoma. The majority of MPNSTs occur in patients between 20-50 years old and for this reason it is hypothesized that are typically an adult life disorder [5]. Some family cancer syndromes are linked to an increased risk of developing soft tissue sarcomas. Evidence supporting this genetic hypothesis includes loss of heterozygosity on chromosome 17p. Actually, mutation of p53 genome at 17p increases the likelihood of cancer, as in neuroblastoma patients [5]. However, the exact cause of this disease is not yet well known.
A 73-year-old woman came to our emergency department with abdominal distension, middle and upper abdominal pain, vomiting and severe vaginal bleeding. The patient has previously referred similar symptoms to the current development and treated by using anti-inflammatory drugs, acid suppression and replacing fluid. An initial Magnetic Resonance Imaging (MRI), that was performed in a private center indicated: Presence of a large, intrauterine mass projecting to the cavity of the uterus and > 50% filtration, which is mainly located in the uterine cavity. In addition, endometrial adenomyosis was observed. Moreover, there was an absence of ascetic collection. No lymph node swelling and no tumor suspicious findings in ovaries were observed in the area of the minor pelvis.
According to these findings it will be probably an endometrial cancer stage Ib papillary serous carcinoma, but the possibility of a sarcoma was not excluded. Due to severe hemorrhage the patient underwent initially a fractional curettage D and C. The histological examination revealed the following: Multiple microscopic sections of the shipped webs (encapsulation of all material) showed, blood clots, among which few webs are recognized in the presence of a malignant, highly cytoplasmic neoplasm. The tumor consists of small to medium-sized neoplastic cells with polymorphic, dark-colored, often giant nuclei, fuzzy cell lines and minimal cytoplasm, arranged on continuous surfaces, often in interconnected bundles. The substrate, in places, shows strong edema, and it also has thick-wall vessels. Areas with increased cell count, alternating with regions of less cellular or myxoidal composition are observed. Mitotic activity is strongly increased (about 20/10 MIC) with frequent presence of atypical nuclei. Immunohistochemical examination was positive for vimemtin, PGP9.5., p53 (100% of tumor cells), CD10, CD99 and CD19, partially positive for S-100, and CD56, while negative for markers actin, CD34, cyclin Dl, p63, Leu 7, NSE, CD117, caldesmon, CD68, HMB45, and Melan A (Figures 1-5). The cell proliferation marker MIB 1 (ki-67) is positive in 95% of the total neoplastic cell population.
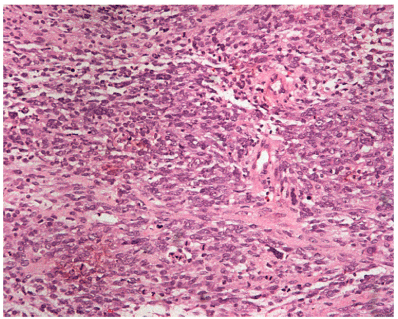
Figure 1. Haematoxylin –eosin stain (magnification x 200)
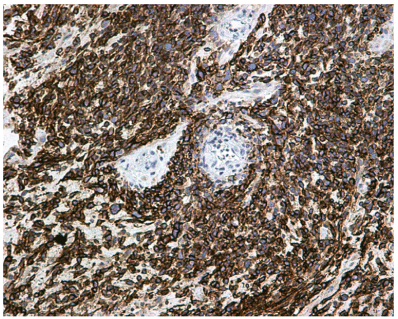
Figure 2. Immunohistochemical positivity for CD56 marker (magnification x 200)
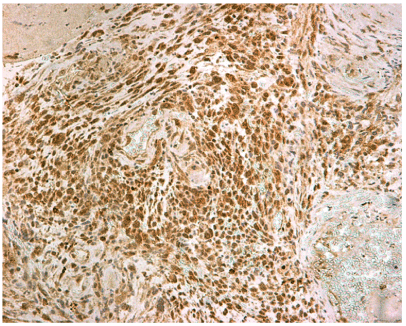
Figure 3. Immunohistochemical positivity for CD19 marker (magnification x 200)
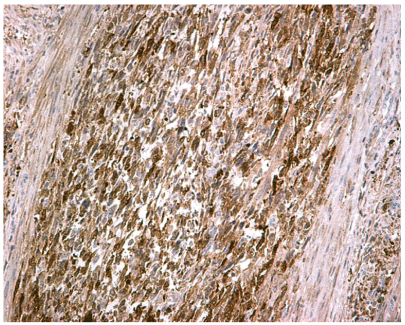
Figure 4. Immunohistochemical positivity for PGP9.5. marker (magnification x 200)
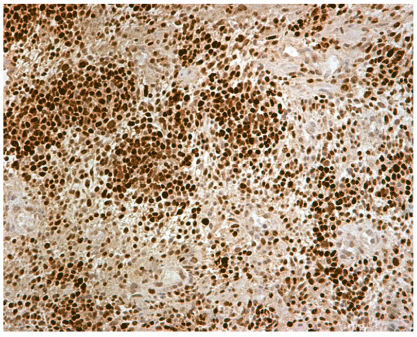
Figure 5. Immunohistochemical positivity for p53 marker (magnification x 200)
The above microscopic and immunohistochemical findings advocate sarcoma, a high degree of malignancy, more compatible with MPNST than with endometrial stromal sarcoma. One week after the curettage, total hysterectomy with both adnexectomy was performed. The specimen was sent for further histopathological examination. The weight and the dimensions of the uterus were 345 g and 11X 11X10.5 cm respectively. Cut section revealed a tumor in the uterine corpus, with the largest dimension being 9,5 cm and on cut surface had fleshy appearance, necrotic areas and hemorrhages. Microscopical and immunhistochemical examination showed the development of a mesenchymal tumor including hypercellular and hypocellular regions. The neoplasm predominately composed of optimal to medium sized cells with hyperchromatic nuclei and pale cytoplasm that showed mainly fascicular growth pattern. These neoplastic cells often tended to concentrate around blood vessels. No heterologous elements were found.
Immunohistochemically, tumor cells were diffuse positive for PGP9.5, p53, CD99, CD19, vimentin and CD10, and in part positive for S100 proteins and CD56, though were negative for a-SMA, desmin, myosin, CD34, CD117, p63 and HMB45. Intraoperative and postoperative course were uneventfully. During the six months a targeted therapy postoperative imagery with MRI of upper and lower abdomen and chest revealed condition (total hysterectomy and both adnexectomy) was performed (Figures 1-5).
An association between MPNST tumor and NF-1 was not exanimated, due to the fact, that special antibody was not available in our Hospital. No signs of local recurrence of the disease (reported neuroendocrine uterine sarcoma) are identified. Lymph nodes remained unchanged. The liver was normal in size, smooth outline and free from focal or diffuse lesions. There was no enlargement of the intrahepatic, extrapulmonary bile ducts or pancreatic duct. Pancreas was normal in size and no focal or diffuse lesions occur. Spleen was of normal size without disturbing its homogeneous density. Kidneys were normal in size and shape and except of a few small simple cysts, had no focal lesions. Moreover, adrenal glands were normal. Abdominal aorta had a diameter within normal limits. There were no overlapping or diffuse collections in the peritoneal cavity or the posterior peritoneal space. No abnormal lymph nodes appeared in the peritoneal cavity or the posterior peritoneal space. Visible periaortal lymph nodes were identified at the level of the renal vessels, up to 7mm in diameter. The bladder had a normal wall. Selective elements were identified in the pulmonary bases, middle lobe and tongue. Minimal fibrous elements were observed in the pulmonary peaks. There were no other significant abnormal findings in pulmonary parenchyma emerge. The medullary lymph nodes were not pathologically sized by imaging criteria. The lymph nodes of the gates, the overlapping areas and the axillary spaces were normal in size. Partially depolarized nodal retraction in the left breast is depicted. A 20 mm diameter nodule was identified in the left lobe of the thyroid gland. The gynecological examination revealed no pathological findings after the follow up after six months. After consideration with the Oncology and Radiotherapy department of targeted radiotherapy of 10 series is recommended.
MPNST are rare malignancies with poor prognosis. In the past, the terms malignant sarcoma, malignant neuralgia, neurogenic sarcoma and neurosarcoma were used to describe this group of tumors which belong to the sarcoma family and are of neuroectodermal origin. They grow from deep soft tissues and are characterized by aggressive biological behavior. MPNSTs can occur as isolated tumors or NF1 [8-10]. People with NF-1 have an increased risk for developing MPNST as these tumors occur on the ground of pre-existing neurinomas that are rarely found in a population that does not have NF-1 [8-10]. Diagnosis is often difficult and delayed, as these tumors are usually asymptomatic at least in the early stages of the disease or cause minimal malfunction [8-10].
In addition, the coexistence of NF1 is not always evaluated, as these patients are treated by physicians of different specialties [8-10]. There are no predetermined criteria for their diagnosis, and there are no widely accepted treatment protocols for the treatment of these patients. MPNST are particularly rare in childhood. At younger ages and potentially associated with a higher incidence of MPNST than the general population of NF1 patients.
The incidence of MPNST in patients with NF1 is estimated between 2 and 5%, while in the general population it reaches 0.001% and therefore [8-10], NF1 sufferers should be closely monitored for the development of MPNST. MPNST are rare in children without neuroimaging, although there are references in the literature to the development of these tumors in 7-year-old patients.
The normal p53 gene will control cell growth and suspend any uncontrolled cell growth in healthy people. Moreover, p53 is not activated in patients with neuromuscular and due to this reason those patients are much more sensitive to growing tumors. Furthermore, despite that a malignant peripheral nerve tumor is not frequent, it is one of the most common soft-tissue sarcomas in pediatric. About half of these cases also occur along with NF-1, which is a genetic mutation on the 17th chromosome that causes tumors along the nervous system. The lifetime risk of NF-1 patients developing MPNST has been estimated at 8-13%, whereas patients with MPNST alone have 0.001% in the general population [11-13]. The NF-1 and MPNST are categorized as autosomal dominant disorders. This means that when someone receives an abnormal gene from one of their parents, they will eventually have this disorder. This individual has a 50/50 chance of transmitting this gene to his offspring. The pedigree title on the right describes this genetic pattern [11-13]. The most convincing test for a patient with a potential neuroimmune tumor is biopsy of the tumor. Magnetic resonance imaging, x-rays, CT scans, and bone abnormalities can help identify tumors and/or possible metastases. Most malignant tumors of the peripheral nerve cavity result from the nerve plexuses that distribute the nerves at the ends - the arms and lumbar plexuses - or the nerves that originate from the trunk [11-13].
MPNSTs found in patients with NF1 are usually diagnosed at a younger age than the general population and are associated with worse prognosis than patients without NF1. A prevalence of male sex has been reported in some studies. Other researchers, on the other hand, did not find any gender prevalence. The primary reason for seeking medical attention in patients with MPNST is usually pain and finding a spinal cord.
Diagnosis of malignant transformation can be difficult even with the use of magnetic resonance imaging or biopsy. Clinicians should suspect possible remission when patients with NF1 experience persistent and intense pain not attributable to another cause, rapid increase in the size of a plexus neurinoma, change in swelling composition with increased stiffness, or even in case of developmental necrosis [14].
Clinicians should suspect possible remission when patients with NF1 experience persistent and intense pain not attributable to another cause, rapid increase in the size of a plexus neurinoma, change in swelling composition with increased stiffness, or even in case of developmental necrosis [14]. Magnetic resonance imaging is not a reliable test for the diagnosis of malignant transformation, but should be used to identify the location of the process, its relation to adjacent tissues and neurovascular structures and the extent of the process or to monitor changes in the size of a neuron. The use of Positron Emission Tomography (18FDG-PET) has been reported as a potentially useful non-invasive method for the detection of malignant transformation of a plexoid neurinoma in patients with NF1 [9,10,14]. However, the distinction between a low degree of MPNST malignancy and benign pancreatic neurinoma is not always possible. As in other studies [9-14], magnetic resonance imaging was used for initial evaluation in our patient. Histologically these tumors are characterized by moderate to significant hypercellularity, nuclear atypia and increased mitotic index. Even the presence of a mitosis can be diagnostic in patients with hypercellularity and nuclear atypia. Other criteria for malignancy are filtration of adjacent tissues by malignant cells, vascular invasion and necrosis. The histological features of these tumors range from benign findings to clear malignancy, and it is often possible to distinguish between high- and low-grade malignancies. However, a significant number of so-called "atypical neurinomas" cannot be classified in any system. These may be locally invasive tumors but are less likely to be associated with metastases [9-14].
MPNST have aggressive behavior and poor prognosis if left untreated. The complete exclusion of the mass is the primary treatment of the disease. Complementary radiotherapy is required for the probability of having a small residual mass. Radiation therapy contributes to local control and retards potential relapse but has little effect on survival. These tumors are relatively resistant to radiotherapy and therefore the complete exclusion of the mass is extremely important in their treatment. The role of chemotherapy is unclear because these tumors are chemo resistant. It is preferred in the case of adults for the treatment of metastatic disease. Drugs such as doxorubicin or its combination with ifosfamide have been used with relative success in the treatment of MPNST [13-14].
However, the effect of chemotherapy appears to be palliative and relieving, as long-term remission with chemotherapy is rare. Also, neoadjuvant chemotherapy can be used to reduce the size of the process and to attempt removal later in large and untreated initial tumors. Most MPNSTs are highly malignant sarcomas with the possibility of local recurrence and distant metastases. 60-70% of these tumors are biologically very aggressive and often develop within two years. The probability of local recurrence ranges from 40-65% while that of metastasis is 40-68%. The most common localization of metastases is the lung followed by the bones. Metastasis to the lymph nodes is rare with a frequency of 0% to 7% in various series. Other metastases are those in the liver, brain, soft tissues, skin, and retrograde. Two of our patients had metastatic lesions in the lung.
In general, MPNSTs have a very poor prognosis compared to other soft-tissue sarcomas. Five-year survival for adults and children with MPNST ranges from 34% to 44%. None of the four patients in our study survived despite extensive exclusion or complementary radiotherapy and/or chemotherapy. Although limb localization appears to be associated with better prognosis than head and neck, localization does not appear to be a determinant of prognosis. However, according to some researchers, patients with distal extremities have a better prognosis. The main predictor is the most extensive process exclusion with a five-year survival rate of 65%. Patients with locally widespread excision or amputation had a longer disease-free survival than those with non-polymorphic exclusion. That is why the purpose of the surgery is to make a complete exception to the healthy. Amputation may be indicated in the event of extensive tumors recurring after appropriate treatment. Prognostic significance is tumor necrosis, high cell count and increased mitotic index. Other adverse prognostic factors are greater tumor volume (> 5 cm), central location, the need for amputation and tumor recurrence. Patients with MPNST without NF1 have a 50-year survival of 50% while in NF-1 territory survival is less favorable with a reported five-year survival of 15% reaching 9% in case of excessive volume 1. In contrast, other researchers found no difference in survival depending on whether or not NF-1 coexisted [13-15].
In conclusion, tumors that grow on NF-1 soil are highly malignant sarcomas with poor survival. Patients with NF-1 should be closely monitored to detect possible change in MPNST in a timely manner. The likelihood of diagnosing an MPNST should be considered when treating a tumor with soft molecules. Despite the high degree of malignancy of these tumors, they may initially appear as benign appearance. The use of imaging methods such as PET-scanning may prove useful in distinguishing MPNSTs from benign neuromas. Clinical and molecular genetics studies are necessary for early detection of patients with NF-1 who are at high risk for developing MPNST. Also, advanced molecular genetics methods may offer the potential for targeted therapies.
Finally, MPNSTs should be tackled with the help of specialist neurologists, geneticists, surgeons and oncologists. in order to have an extensive experience in the area to ensure the best possible treatment. Patients can also benefit greatly from being treated by medical teams specializing in soft tissue sarcomas. Complete surgical excision of the tumor significantly increases disease-free survival, while the role of chemotherapy is unclear, and its efficacy is still not clearly defined as it has provided only marginal survival benefit [16]. In addition, all patients should receive complementary radiotherapy [17]. However, radiation was not found to be a prognostic factor for overall survival [16].
Our case report presentation is associated to limitations as previously published papers with similar referred cases (only nine cases) with the same histological findings and two existing in follow up alive were published. In the referred cases, the majority were located in the uterine cervix [18] while just another case is mentioned in literature like in our clinical case arising from uteri corpus [11-15] and that patient did not have that prolonged free disease time. There is no reported established treatment. In conclusion, due to these mentioned reasons this case is very rare and, we describe clinicopathologic features and postoperative course of uterine corpus MPNST and is likely to be of great interest to the scientists, researchers and clinicians.
- Wanebo JE, Malik JM, VandenBerg SR, Wanebo HJ, Driesen N, et al. (1993) Malignant peripheral nerve sheath tumors. A clinicopathologic study of 28 cases. Cancer 71: 1247-1253. [Crossref]
- Hasegawa K, Udagawa Y, Fukasawa I (2018) Spindle cell sarcoma of the uterine corpus. Austin J Obstet Gynecol 5: 1089.
- Stucky CC, Johnson KN, Gray RJ, Pockaj BA, Ocal IT, et al. (2012) Malignant peripheral nerve sheath tumors (MPNST): The Mayo Clinic experience. Ann Surg Oncol 19: 878-885.
- James AW, Shurell E, Singh A, Dry SM, Eilber FC (2016) Malignant peripheral nerve sheath tumor. Surg Oncol Clin N Am 25: 789-802.
- Miettinen MM, Antonescu CR, Fletcher CDM, Kim A, Lazar AJ, et al. (2017) Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol 67: 1-10. [Crossref]
- Kim ET, Namgung H, Shin HD, Lee SI, Kwon JE, et al. (2012) Oncologic manifestations of neurofibromatosis type 1 in Korea. J Korean Surg Soc 82: 205-210. [Crossref]
- Kroep JR, Ouali M, Gelderblom H, Le Cesne A, Dekker TJ, et al. First-line chemotherapy for malignant peripheral nerve sheath tumor (MPNST) versus other histological soft tissue sarcoma subtypes and as a prognostic factor for MPNST: an EORTC soft tissue and bone sarcoma group study. Ann Oncol 22: 207-214. [Crossref]
- Inoue T, Kuwashiro M, Misago N, Narisawa Y (2014) Superficial malignant peripheral nerve sheath tumor arising from diffuse neurofibroma in a neurofibromatosis type 1 patient. J Dermatol 41: 631-633. [Crossref]
- Hajari ARS, Tilve AG, Kulkarni JN, Bharat R (2015) Malignant peripheral nerve sheath tumor of the uterine corpus presenting as a huge abdominal neoplasm. J Cancer Res Ther 11: 1023. [Crossref]
- Wells AE, Mallen AM, Bui MM, Reed DR, Apte SM (2019) NTRK-1 fusion in endocervical fibroblastic malignant peripheral nerve sheath tumor marking eligibility for larotrectinib therapy: A case report. Gynecol Oncol Rep 28: 141-144. [Crossref]
- Fadare O (2006) Uncommon sarcomas of the uterine cervix: a review of selected entities. Diagn Pathol 1: 30. [Crossref]
- Rodriguez AO, Truskinovsky AM, Kasrazadeh M, Leiserowitz GS (2006) Case report: Malignant peripheral nerve sheath tumor of the uterine cervix treated with radical vaginal trachelectomy. Gynecol Oncol 100: 201-204. [Crossref]
- Miguchi M, Takakura Y, Egi H, Hinoi T, Adachi T, et al. Malignant peripheral nerve sheath tumor arising from the greater omentum: case report. World J Surg Oncol 9: 33. [Crossref]
- de A Focchi GR, Cuatrecasas M, Prat J (2007) Malignant peripheral nerve sheath tumor of the uterine corpus: a case report. Int J Gynecol Pathol 26: 437-440. [Crossref]
- Houreih MA, Eyden B, Deolekar M, Banerjee S (2007) A case of fibroblastic low-grade malignant peripheral nerve sheath tumor--a true neurofibrosarcoma. Ultrastruct Pathol 31: 347-356. [Crossref]
- Farid M, Demicco EG, Garcia R, Ahn L, Merola PR, et al. (2014) Malignant peripheral nerve sheath tumors. Oncologist 19: 193-201.
- Kahn J, Gillespie A, Tsokos M, Ondos J, Dombi E, et al. (2014) Radiation therapy in management of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Front Oncol 4: 324. [Crossref]
- Trisal M, Rana S, Sultan B, Jetle S (2019) Primary malignant schwannoma of the uterine cervix: Unsual site posing a diagnostic challenge. Scholar J App Med Sci 7: 3430-3432.