Autosomal recessive congenital ichthyosis (ARCI) is a rare hereditary disorder of cornification. ARCI is a congenital recessive skin disorder characterized by generalized scaling and hyperkeratosis. Mutations in the transglutaminase-1 (TGM1) gene, which encodes for the epidermal enzyme transglutaminase-1, are one of the causes of ARCI. The transglutaminase 1enzyme, is critical for the assembly of the cornified cell envelope in terminally differentiating keratinocytes. In this study a nine-year old boy who presented with ARCI. The whole exome sequencing WES of transglutaminase-1 gene was investigated. The patient harbored a homozygeous mutation of C.1165T>C transition located in exon8 of TGM1 gene, resulting in the substitution of argenine by serine at amino acid position 389. The parents and one of the sibs were hetrozygeous for the variant and one was homozygeous for wild allele. The mutated allele was not found in controls. This mutation localized in this study correspond to the core catalytic core domain of enzyme, based on provean algorithm, the variant at position level was predicted to decrease TGM1 Enzyme activity by producing an unstable protein.
Transglutaminase-1 Gene, autosomal recessive congenital ichthyosis, hyperkeratosis
Autosomal recessive congenital ichthyosis (ARCI) is a clinically heterogeneous group cornification of disorder of the skin. With an incidence of an approximately 1 in 200,00 birth [1-3]. ARCI is called congenital, since it appears at birth or shortly thereafter. The major phenotypic subtype of ARCI includes lamellar ichthyosis [LI; (OMIM; 242300)] and non-bullous congenital ichthyosis form erythroderma [NBCIE; (OMIM; 242100)] [3,4].
Large darke, plate-like cutaneous scales with minimal erythema is a feature of LI patients, whereas, in NBCEI patient’s erythroderma with overlying white scales is a common feature of the disease [5-7].
While ARCI is genetically heterogeneous, with at least twelve genes known to be associated with ARCI (TGM1, ABCA12 Ich thin (known as NIPAL4), ALOX12B, ALOXE3, LIPIN, CYP4F22, ST14, CASP14, SDR9C7, SULT2B1, CERS3 and PNPLA1) [2,3,7-17]. The most common forms, approximately 30% of the heritability of ARCI is explained by mutation in the gene, encoding keratinocyte transglutaminase (TGM1: 190195) an enzyme involved in the formation of the cornfield envelope [18,19].
TGM1 gene is located on chromosome 14q11.2, codant for a protein with a molecular weight of 89 KD. TgM1 has 14095 bp, including 2454 bp of coding sequence, and spans15 exons (Gene bank NM-000359.2) [20,21]. TGM1 has been associated with the development of the cornfield cell envelope (CCE). Human epidermal scale and nails of ARCI patients with TGM1 mutations have shown a structurally perturbed or attenuated CEE [22]. CEE is a physical and water impermeable barrier that replaces the plasma membrane in mature skin cells. ARCI patients with TGM1 mutations have shown a structurally perturbed or attenuated CCE [18].
In this study exome sequencing applied for an Iranian ichthyosis patient, and a novel homozygous causing variant in TGM1 was identified.
A nine-year-old boy suffering from ichthyosis was the proband of the study. Dry, rough scaly skin all over the body was started at birth. Clinical features include: dark areas over the elbows and knees, scaring deformity of the hands, depressed nasal bridge, non-developed ears are other signs of the disease in the proband, prominent erythroderma and fine white, superficial semi adherent scales nail dystrophy, subungual hyperkeratosis, scalp involvement and loss of eyebrows was observed in the proband in clinical examination. Proband was the yield of first cuisine marriage, the two other children of the parents were normal, and the mother has been experienced a miscarriage.
Whole exome sequencing WES has been used extensively to diagnosis novel disease and find novel causative mutations for known disease phenotypes. In this issue WES is investigated. After taking informed consent, 5 ml peripheral blood sample were drowned from patient and other members of the family. DNA extracted from peripheral blood lymphocytes by BIOGENT, Korean DNA extraction kit for WES using the Maxwell RSC instrument (standard instrument protocol). DNA fragments containing targeted coding sequences were captured using the seqcapEZ Med exome target enrichment kit and sequenced on the illumine Hiss equation 2500 platform. The resulting sequence was analyzed for single nucleotide variants and small insertion and deletions differing from the reference genome (human genome 19 (HG 19). In this study the mutations affected the predominant gene that causes ARCI encoding keratinocyte transglutaminase (TGM1) investigated.
Clinical characterization of affected individual included a dry rough scaly skin all the body with dark area over the elbow diagnosed as nonbullous congenital ichthyosis form erythroderma (NCIE). A form of the autosomal recessive congenital ichthyosis ARCI. ARCI is a heterogeneous group of congenital keratinization disorders [15,23,24]. The most common Causes of ARCI are the mutations in TGM1, that encodes Transaminase-1 enzyme [18].
To date, more than 115 various mutations in the TGM1 gene in 234 unrelated patients from different racial/ethnic backgrounds have been reported and the common type of the mutation is missense. The most common reported mutations in north America and Norouch patients was the c.877-2 A>G [18].
In this paper, the clinical and molecular aspects of ARCI have been investigated in patients from Iran. The proband showed the typical features of ARCI.
Molecular finding showed a novel missense variant c.1165C>T; p. Arg389Ser in TGM1 in proband. A novel pathogenic variant in exon 8. Sanger sequencing was performed for DNA samples of other members of the family. The variant was not found in 1000 genomes project, NCBI db SNP human build, NCBI clinvar db, exome aggregation consortium v.0.3.1. (Exac), Human gene mutation db (HGMD), and NHLBI exome sequencing project.
Homozygous state of c.1165 C>T was observed in the proband, parents and one of the sibs were heterozygous for the variant and one sib was homozygous for wild allele (Figure 1). The mutated allele was not found in 100 controls. In silico analysis was done by mutation taster 2.0 and FATHMM MKL coding prediction tools and the effect of the variant on protein was evaluated by Poly-phen 2.0, SIFT and PROVEN online soft- wares. Variant calling was done according to American College of Medical Genetics (ACMG) guidelines. The outcome effect is substitution of Arginine by serine in position 389 of the protein, a highly conserved position. Based on conservation scores this novel variant is probably damaging. Substitution of Arginine by serine can cause great effect on protein configuration by loss of one positive charge, because serine is a neutral amino acid, in addition serine is smaller than
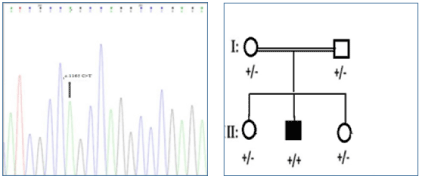
Figure 1. A, The Sanger sequencing result of proband. The variant (c.1165 C>T; p.R389C) is shown by an arrow. The affected member (II-2) Is homozygous for the variant, and both parents are heterozygous. B, Family pedigree showing one affected boy. Proband is shown by an arrow.
Arginine in size. It seems that Arginine in position 389 play an important role, two missense mutations in this site have been reported by Akima (p.R389H) and Shevchenkov (p. R389P) in ARCI patients [24,25]. Also, the variant p.R389H in compound heterozygous state with p.R307W in TGM1 has been reported as ARCI causing [26].
In protein level, p.R389C decreases the TGM1 enzyme activity by producing an unstable protein, based on PROVEAN prediction software [18,27]. From the mutational analysis investigated by WES, the proband was confirmed to be a homozygeous c>t transition at position 1165 in the exon 8 region with genotype Missence.Arg of TGM1 gene. The origin wild type residue and novely introduced mutant residue often differ in these properties. The mutant residue (ser.) is smaller than the wild-type residue (Arg.) that the wild-type residue charge is positive while the mutant residue charge is neutral. Sequential arrangement of the domain structure of transglutaminase reveal that the novel mutation localized in this study correspond to the core catalytic domain of this enzyme. The core catalytic domain started at amino acid positioned in 224 to end 577 area. The previous study defined some other mutations in the exone 8 (Table 1) were in the catalytic core domain [18,28]. Definitive and interpretation of the effects of these mutations on the catalytic functions of the TGM1 protein cannot be provided, owing to lack of good models. However, based on provean algoritm, the variant (p. Arg389Cys) at the protein level was predicted to decrease TGM1 enzyme activity by producing an unstable protein.
Table 1. The TGMA1 gene mutations in the exone 8 associated with core catalytic domain.
Nucleotide change NM
000359.2 |
Amino acid change NP
000350 |
References |
C.1213c>A |
p.His405Asn |
Herman et al. |
C.1223-1227del.ACACA |
p.Asp408valfs×21 |
Herman et al. |
C.1261A>G |
p.Met421Val |
Herman et al. |
C.1165C.T |
p.Arg389His |
Herman et al. |
|
C.G>T |
p.Arg395Leu |
Laiho et al. |
P.Arg389Pro |
Laiho et al. |
P.Gly392Asp |
Laiho et al. |
P.Arg395Leu |
Laiho et al. |
C.918C>G |
p.Asp306Glu |
Pigget al. |
C.1094A>G |
p.Tyr365Cys. |
Pigget al. |
C.1163T>C |
p.Leu.388Pro. |
Pigget al. |
C.1389A>T |
p.Gln463His |
Pigget al. |
C.1483A>T |
p.Ile480Phe |
Pigget al. |
C.1165C>T |
p.Arg389Ser |
This report |
In conclusion our study revealed one mutation in the TGM1 gene in an Iranian patient. This mutation is a novel variant missence (c.1165C>T) and may also be prevalent among Iranian populations. The results are useful for genetic counseling in the pedigree and applicable for preventing disease recurrence via preimplantation genetic diagnosis.
- Shawky RM, Sayed NS, Elhawary NA (2004) Mutations in transglutaminase 1 gene in autosomal recessive congenital ichthyosis in Egyptian families. Dis Markers 20: 325-332. [Crossref]
- Esperón-Moldes U, Ginarte Val M, Rodríguez-Pazos L, Fachal L, Azaña JM, et al. (2019) Novel and Recurrent PNPLA1 Mutations in Spanish Patients with Autosomal Recessive Congenital Ichthyosis; Evidence of a Founder Effect. Acta Derm Venereol 99: 894-898. [Crossref]
- Karim N, Ullah A, Murtaza G, Naeem M (2019) Molecular Genetic Study of a Large Inbred Pakistani Family Affected with Autosomal Recessive Congenital Ichthyosis Through Whole Exome Sequencing. Genet Test Mol Biomarkers 23: 428-432. [Crossref]
- Krieg P, Dick A, Latzko S, Rosenberger S, Meyer J, et al. (2020) Conditional Alox12b knockout: degradation of the corneocyte lipid envelope in a mouse model of autosomal recessive congenital ichthyoses. J Invest Dermatol 140: 249-253. [Crossref]
- Esperon-Moldes US, Pardo-Seco J, Montalvan-Suarez M, Fachal L, Ginarte M, et al. (2019) Biogeographical origin and timing of the founder ichthyosis TGM1 c. 1187G> A mutation in an isolated Ecuadorian population. Scientific Reports 9: 7175.
- Hartley IR, Nunes JCB, Lodish M, Stratakis CA (2019) Metabolism. Cushing disease in a patient with nonbullous congenital ichthyosiform erythroderma: lessons in avoiding glucocorticoids in ichthyosis. J Pediatr Endocrinol Metab 32: 911-914. [Crossref]
- Williams ML, Elias PM (1985) Heterogeneity in autosomal recessive ichthyosis: clinical and biochemical differentiation of lamellar ichthyosis and nonbullous congenital ichthyosiform erythroderma. Arch Dermatol 121: 477-488. [Crossref]
- Ballin N, Hotz A, Bourrat E, Kusel J, Oji V, et al. (2019) Genetical, clinical and functional analysis of a large international cohort of patients with autosomal recessive congenital ichthyosis due to mutations in NIPAL4. Hum Mutat 40: 2318-2333. [Crossref]
- van Leersum F, Seyger M, Theunissen T, Bongers EMHF, Steijlen PM, et al. (2019) Recessive mosaicism in ABCA 12 causes blaschkoid congenital ichthyosiform erythroderma. Br J Dermatol 182: 208-211. [Crossref]
- Borská R, Pinková B, Réblová K, Bucková H, Kopecková L, et al. (2019) Inherited ichthyoses: molecular causes of the disease in Czech patients. Orphanet J Rare Dis 14: 92. [Crossref]
- Seidl-Philipp M, Schossig AS, Moosbrugger-Martinz V, Zschocke J, Schmuth M, et al. (2019) Impaired epidermal barrier in autosomal recessive congenital ichthyosis (ARCI) caused by missense mutations in SDR9C7 in two Austrian sisters. J Dtsch Dermatol Ges 17: 742-745. [Crossref]
- Briand A, Cochet-Faivre N, Reyes-Gomez E, Jaraud-Darnault A, Tiret L, et al. (2019) NIPAL4 deletion identified in an American Bully with autosomal recessive congenital ichthyosis and response to topical therapy. Vet Med Sci 5: 112-117. [Crossref]
- Simpson J, Martinez-Quiepo M, Onoufriadis A, Tso S, Glass E, et al. (2019) Genotype-phenotype correlation in a large English cohort of autosomal recessive ichthyosis. Br J Dermatol. [Crossref]
- Karim N, Durbin-Johnson B, Rocke DM, Salemi M, Phinney BS, et al. (2019) Proteomic manifestations of genetic defects in autosomal recessive congenital ichthyosis. J Proteomics 201: 104-109. [Crossref]
- Lima Cunha D, Alakloby OM, Gruber R, Kakar N, Ahmad J, et al. (2019) Unknown mutations and genotype/phenotype correlations of autosomal recessive congenital ichthyosis in patients from Saudi Arabia and Pakistan. Mol Genet Genomic Med 7: e539. [Crossref]
- Heinz L, Kim G-J, Marrakchi S, Christiansen J, Turki H, et al. (2017) Mutations in SULT2B1 cause autosomal-recessive congenital ichthyosis in humans. Am J Hum Genet 100: 926-939. [Crossref]
- Radner FP, Marrakchi S, Kirchmeier P, Kim G-J, Ribierre F, et al. (2013) Mutations in CERS3 cause autosomal recessive congenital ichthyosis in humans. PLoS Genet 9: e1003536. [Crossref]
- Herman ML, Farasat S, Steinbach PJ, Wei MH, Toure O, Fleckman P, et al. (2009) Transglutaminase-1 gene mutations in autosomal recessive congenital ichthyosis: summary of mutations (including 23 novel) and modeling of TGase-1. Hum Mutat 30: 537-547. [Crossref]
- Boeshans KM, Mueser TC, Ahvazi BJ (2007) A three-dimensional model of the human transglutaminase 1: insights into the understanding of lamellar ichthyosis. J Mol Model 13: 233-246. [Crossref]
- Kim I-G, McBride O, Wang M, Kim S, Idler W, et al. (1992) Structure and organization of the human transglutaminase 1 gene. The J Biol Chem 267: 7710-7717.
- Yamanishi K, Inazawa J, Liew F, Nonomura K, Ariyama T, et al. (1992) Structure of the gene for human transglutaminase 1. J Biol Chem 267: 17858-17863. [Crossref]
- Rice RH, Crumrine D, Uchida Y, Gruber R, Elias PM (2005) Structural changes in epidermal scale and appendages as indicators of defective TGM1 activity. Arch Dermatol Res 297: 127-133. [Crossref]
- Sathishkumar D, Peter D, Pulimood S, Wiegmann H, Valentin F, et al. (2018) Bathing Suit Variant of Autosomal Recessive Congenital Ichthyosis (ARCI) in Two Indian Patients. Case Rep Dermatol Med 2018: 3140473. [Crossref]
- Shevchenko YO, Compton JG, Toro JR, DiGiovanna JJ, Bale SJ (2000) Splice-site mutation in TGM1 in congenital recessive ichthyosis in American families: molecular, genetic, genealogic, and clinical studies. Hum Genet 106: 492-499. [Crossref]
- Esposito G, De Falco F, Neri I, Graziano C, Toschi B, et al. (2013) Different TGM1 mutation spectra in Italian and Portuguese patients with autosomal recessive congenital ichthyosis: evidence of founder effects in Portugal. Br J Dermatol 168: 1364-1367. [Crossref]
- Muramatsu S, Suga Y, Kon J, Matsuba S, Hashimoto Y, et al. (2004) A Japanese patient with a mild form of lamellar ichthyosis harbouring two missense mutations in the core domain of the transglutaminase 1 gene. Br J Dermatol 150: 390-392. [Crossref]
- Russell LJ, DiGiovanna JJ, Rogers GR, Steinert PM, Hashem N, et al. (1995) Mutations in the gene for transglutaminase 1 in autosomal recessive lamellar ichthyosis. Nat Genet 9: 279-283. [Crossref]
- Laiho E, Ignatius J, Mikkola H, Yee VC, Teller DC, et al. (1997) Transglutaminase 1 mutations in autosomal recessive congenital ichthyosis: private and recurrent mutations in an isolated population. Am J Hum Genet 61: 529-538. [Crossref]