Objectives: Inherited genetic defects are the most common causes of autosomal recessive nonsyndromic hearing loss (ARNSHL), affecting millions of people worldwide. Identification and specific molecular diagnosis of the pathogenic genes or loci would facilitate basic research of ARNSHL and its clinical prevention and treatment.
Materials and methods: We identified the causal deafness gene of a Chinese family with ARNSHL using targeted massively parallel sequencing. We also used DNA from 51 Chinese familial patients with ARNSHL and 60 ethnicity-matched normal controls to perform extended variants analysis.
Results: We detected a novel compound heterozygous mutation, c. 10419_10423delCAGCT and a previously reported mutation c. 6956+9C>G, in MYO15A gene in the proband. Both mutations co-segregated with hearing loss in the Chinese family in our study and were absent in the 51 index patients and 60 ethnicity-matched normal controls.
Conclusion: We identified a novel c. 10419_10423delCAGCT frameshift mutation, which results in a truncated MYO15A protein that lacks part of the second FERM domain and PDZ-ligand at the C terminus. Our results demonstrate that the novel c. 10419_10423delCAGCT mutation, in compound heterozygosity with the previously reported c. 6956+9C>G splicing site mutation, is the underlying cause of ARNSHL in this Chinese family. Our study extends the mutation spectrum of the MYO15A gene for establishing a better understanding of DFNB3.
Abbreviations
ARNSHL: Autosomal Recessive Nonsyndromic Hearing Loss; NSHL: Nonsyndromic Hearing Loss; CT: Computed Tomography; MRI: Magnetic Resonance Imaging; ABR: Auditory Brainstem Response; DPOAE: Distortion Products Otoacoustic Emissions; PCR: Polymerase Chain Reaction; NCBI: National Center for Biotechnology Information; SNPs: Single Nucleotide Polymorphisms; ATP: Adenosine Triphos-Phate
autosomal recessive sensorineural hearing loss, targeted massively parallel sequencing, MYO15A, gene mutation
Hearing impairment is a common sensorineural disorder with a prevalence of over one in a thousand children, and more than 50% of childhood hearing impairment is caused by genetic factors [1,2]. Non-syndromic hearing loss (NSHL) without other associated clinical features, accounts for approximately 70% of genetic cases [3]. Autosomal recessive NSHL (ARNSHL) accounts for up to 80% of cases of inherited hearing loss [4]. To date, more than 60 genes and 100 genetic loci have been identified for ARNSHL (http://hereditaryhearingloss.org; http://deafnessvariationdatabase.org/). Mutations in GJB2, SLC26A4, MYO15A and OTOF genes are most commonly identified in ARNSHL and patients with mutations in these genes may gain satisfactory auditory performance after cochlear implantation [5,6]. Therefore, specific genetic diagnosis and mutations spectrum expansion plays an important role in clinical management, prognosis evaluation and counselling for ARNSHL families.
Autosomal Recessive Deafness 3 (DFNB3) caused by mutation in MYO15A is a common congenital severe-to-profound ARNSHL. MYO15A (NM_016239) has 66 exons distributed on chromosomal locus 17p11.2 in human [7]. Myosin XVa, the protein encoded by MYO15A, is composed of 3,530 amino acids, and is an unconventional myosin and critical for stereocilia elongation and staircase-like pattern formation of stereocilia in cochlear and vestibular hair cells. Myosin XVa is localized at the tip of stereocilia and is a motor protein that uses adenosine triphos-phate (ATP) to move along actin filaments and participate in generating mechano-electrical transduction at the stereociliary tips [8,9]. It has been reported that MYO15A deficient mice (shaker 2) are devoid of tiplinks between there abnormally short stereocilia [10,11]. Mutations in MYO15A/Myo15a result in hearing loss in both humans (DFNB3) and mice (shaker 2), suggesting the defective myosin XVa caused by genetic mutations might lead to a disruption of the mechano-electrical transduction machinery [12-15]. Several mutations in MYO15A causing DFNB3 hearing loss have been identified by linkage analysis in individuals from different countries, such as Pakistan, Turkey, Iran, China, Korea and Brazil [13-19]. Recently, targeted resequencing and massively parallel sequencing (MPS) has been introduced and proved as a more effective alternative approach to traditional methods, owning to its ability to perform parallel sequencing of billions of nucleotides at high speed and a low cost [20-22]. In this study, we used MPS to identify a novel compound heterozygous mutation of MYO15A (c.10419_10423delCAGCT) that in conjunction with a known mutation (c.6956+9C>G) causes ARNSHL in a Chinese family. This novel variant expands the MYO15A mutation spectrum in the Chinese population.
Subjects
A three-generation consanguineous Chinese family with ARNSHL was recruited from Jiangsu province, China (Figure 1). Three members of the family were evaluated by pure tone audiometry (PTA) at frequencies of 125, 250, 500, 1000, 2000, 4000, and 8000 Hz. Besides PTA, the proband underwent tympanometry, auditory brainstem response (ABR), distortion products otoacoustic emissions (DPOAE) and imaging examination performed by computed tomography (CT) and Magnetic resonance imaging (MRI). The patient’s parents were interviewed for a detailed medical history, family history, mother’s health condition during pregnancy and patient’ s clinical history (possible head or brain injury, infection and the usage of medicine including aminoglycoside antibiotics). To screen for candidate mutations, we used 51 affected DNA samples from patients presenting with ARNSHL and in whom mutations of SLC26A4 had been previously excluded. 60 ethnically-matched unrelated subjects with normal hearing were enrolled as controls. Written informed consent was obtained from the participants or guardians, and this study protocol was performed in accordance with institutional bioethics guidelines and was approved by the Research and Ethics Committee of Drum Tower Hospital affiliated with Nanjing University Medical School (201601502).
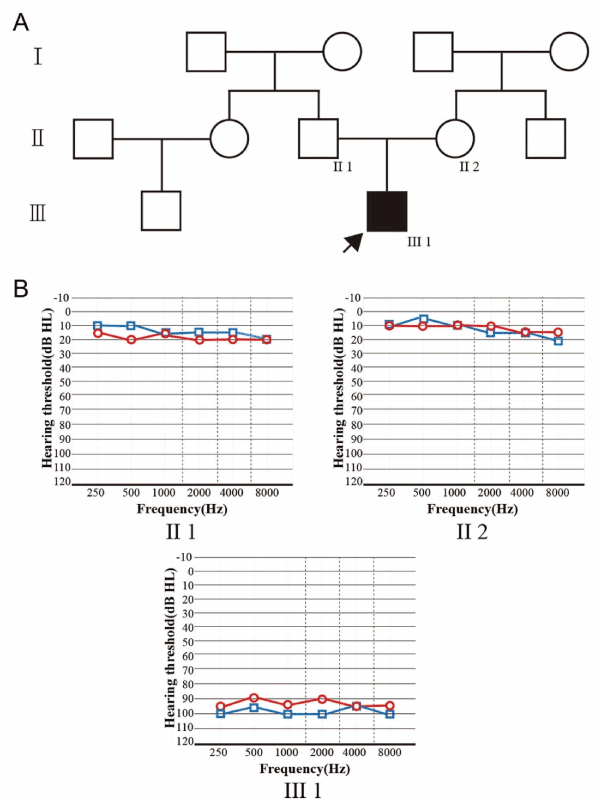
Figure 1.(A) Pedigree of the family with ARNSHL. Darkened symbol with Arrow indicates the proband. (B) Bilateral pure tone audiograms from individuals of II1, II2, III3. Red circles in the audiograms represent right air conduction thresholds and blue squares represent left conduction thresholds.
Molecular genetic testing
Genomic DNA of all family members was extracted from peripheral blood using the Blood DNA kit (TIANGEN BIOTECH, Beijing, China). 1ug of purified gDNA was fragmented to 200–300 base pairs using an ultrasonoscope. End-repair, adenylation and adapter ligation were performed for library preparation. A customized capture array (Roche-NimbleGen) contains 127 deafness genes GJB2, SLC26A4, GJB6, MYO15A, CDH-23, MT-RNR1 et al. [23]. The chondrogene was designed to capture all their exons and their flanking introns(±10bp) sequences. After removing the low-quality reads from the primary data according a local algorithm, data analysis and bioinformatics processing were performed based on the reference sequences of the National Centre for Biotechnology Information (NCBI)37/hg19 assembly of the human genome using the Burrows–Wheeler Aligner (BWA) program.
Variants were annotated with a BGI (Beijing Genomics Institute, Beijing, China) developed annotation pipeline and the frequencies of previously identified SNPs (single nucleotide polymorphisms) were determined using the NCBI dbSNP 135,1000 human Genomes. After identifying the potential mutations by filtering against multiple databases, Sanger sequencing was used to confirm the variant by analyzing the DNA sequences from the proband, his parents, 51 Chinese familial patients with ARNSHL and 60 normal hearing controls.
Clinical presentation
The Chinese family includes a proband, two unaffected parents and six unaffected members (Figure 1A). The proband presented bilateral profound sensorineural hearing loss with thresholds over 90 dBHL was revealed by PTA (Figure 1B) and his bilateral hearing cannot be improved with ear hearing aids. ABR showed that no representative wave could be initiated under 105 dB (the highest intensity) and DPOAE showed no representative response from 1 to 4 kHz. The otoscopic examination revealed normal external auditory canal and both tympanic membranes. The tympanometric results were normal. The proband showed no vestibular symptoms and had normal walking age of 14 months. CT and MRI scan revealed no inner ear malformations. The parents reported no history of previous deafness-causing illness, such as meningitis, trauma of the proband and the pregnancy and delivery were normal. The parents showed normal hearing (Figure 1B). Otoscopy and full physical examination also showed no sign of systemic illness or dysmorphic features. Lastly, other members of the family do not have hearing dysfunction.
Targeted massively parallel sequencing
We performed targeted MPS of all exons and exon–intron boundaries for 127 deafness genes in the proband. After preliminary trimming and alignment to the human genome, we identified 269 variants in the coding regions and introns that might affect splicing. These variants were filtered against allele frequencies from the databases NCBI dbSNP, Deafness Variation Databases of the University of Iowa and The Human Gene Mutation Database. As a result, we focused on genes with homozygous or compound heterozygous variants based on the assumption of an autosomal recessive mode of inheritance in this family. Subsequently, a novel compound heterozygous mutation, c. 6956+9C>G and c. 10419_10423delCAGCT, in MYO15A was detected in the proband in our study (Figure 2B and 2D).
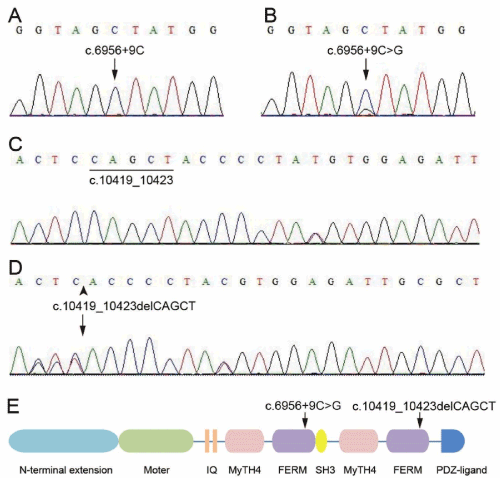
Figure 2.(A) Normal allele c.6956+9C of MYO15A. (B) Heterozygous inheritance of the MYO15A c. 6956+9C>G missense mutation was detected using Sanger sequencing in the proband and his father. (C) Normal allele c.10419_10423 of MYO15A. (D) Heterozygous inheritance of the c.10419_10423delCAGCT deletion of the MYO15A detected using Sanger sequencing in the proband and his mother. (E) Schematic of myosin XVA. Black arrows indicate the novel compound mutation c. 6956+9C>G and c. 10419_10423delCAGCT identified in this study.
Identification of pathogenic mutation
The compound heterozygous mutation of MYO15A was confirmed by Sanger sequencing. The heterozygous c. 6956+9C>G variant was identified in his unaffected father (Figure 2A and 2B) and the heterozygous c. 10419_10423delCAGCT variant was identified in his unaffected mother (Figure 2C and 2D). MYO15A c. 6956+9C>G variant has been detected, affecting intron splicing while c. 10419_10423delCAGCT variant has not been reported before, which was predicted to lead to a shift in the reading frame at amino acid position 3473 and a premature stop codon (p.S3473Sfs*43) by Mutation Taster [24,25]. However, the variants were absent in 51 Chinese familial patients with ARNSHL and 60 ethnically-matched unrelated controls (Figure 2A and 2C), and are likely the disease-causing mutations in the proband in our study.
Despite the linkage analysis and candidate gene screening in previous reports, a large quantity of ARNSHL remains genetically unexplained. Current advancements in TGE and MPS technologies provide more opportunities to identify causative DNA mutations of rare deafness genes. In the present study, novel compound heterozygous mutations, c.10419_10423delCAGCT and c.6956+9C>G (NM_016239), were identified in MYO15A gene.
Several mutations in MYO15A gene have previously been identified in DFNB3 autosomal recessive hearing loss from diverse populations in the world [14,15,26-28]. It is well-known that the MYO15A protein is critical for stereocilia development and elongation through delivery of the cytoskeletal protein whirlin to the tips of the hair cells stereocilia [9]. MYO15A has been showed involving in hair bundle staircase formation, which is essential to sound detection [9].
Human MYO15A consists of an N-terminal domain, motor domain, IQ motifs, MyTH4, FERM, SH3 domains and a PDZ-binding motif at the C terminus [16,26] (Figure 2E). The intronic variants, c.6956+9C>G mutation is predicted to lie in the first FERM domain of MYO15A and have previously been detected in congenital hearing loss [25] (Figure 2E). Band 4.1 fezrin/radixin/moesin (FERM) domains are found in a group of homologous and highly conserved proteins including the same band superfamily. Proteins with FERM domains may function as cross-links between the membrane and the actin cytoskeleton by interacting with the cytoplasmic domains of integral membrane proteins including CD44, CD43 and ICAM-2 [29]. Therefore, c.6956+9C>G mutation affecting the FERM domain, interrupting the multiple integral proteins binding are required for stereocilia development and function.
We identified a novel c.10419_10423delCAGCT variant in exon 62 of MYO15A. The variant was located in the second FERM domain. It was predicted to cause a reading frame shift and a premature stop codon (p.S3473Sfs*43), resulting in a truncated protein missing part of the second FERM domain and PDZ-ligand in the C terminal of MYO15A (Figure 2E). The second FERM region with the second MyTH4 domain is involved in microtubule binding and the formation of the MYO15A-whirlin-Eps8 complex [30,31]. Indeed, the formation of the MYO15A-whirlin-Eps8 complex, which is necessary for stereocilia elongation and sound detecting might interfered by the c.10419_10423delCAGCT variant in the MYO15A gene. Consistent with our finding, other mutations in the second FERM domain and PDZ-ligand of MYO15A have also been identified in DFNB3 patients with severe to profound sensorineural deafness [13,32]. Therefore, the novel compound heterozygous MYO15A mutations (c.10419_10423delCAGCT and c.6956+9C>G) found in the Chinese family may produce defective MYO15A protein that fails to cross-link the integral membrane proteins with the actin cytoskeleton, thus resulting in stereocilia elongation defects and bilateral profound sensorineural hearing loss.
Comparing to the common deafness genes such as GJB2 and SLC26A4, MYO15A is not routinely included in genetic diagnostic tests in Chinese patients due to its less common occurrence and large gene size. Our study not only identified the novel compound heterozygous mutations (c.10419_10423delCAGCT and c.6956+9C>G) that expand the mutation spectrum of MYO15A in the Chinese population, but also highlighted the value of molecular diagnostic techniques including TGE and MPS in uncovering novel and rare deafness mutations.
Ethics approval and consent to participate
Informed consent had been provided by all participants and the parents of all minors in the study, and the study protocol was performed in accordance with institutional bioethics guidelines and was approved by the Research and Ethics Committee of Drum Tower Hospital affiliated with Nanjing University Medical School (201601502).
The study was supported by General Program from Natural Science Foundation of China to Gao Xia (No. 81771019, 81570921) and Chen Jie (No. 81870721). Moreover, the authors would like to thank the participants and Core Medical and Genetics laboratory of Drum tower Hospital affiliated with Nanjing University Medical School.
- Mahdieh N, Shirkavand A, Rabbani B, Tekin M, Akbari B, et al. (2012) Screening of OTOF mutations in Iran: a novel mutation and review. Int J Pediatr Otorhinolaryngol 76: 1610-1615. [Crossref]
- Duman D, Tekin M (2012) Autosomal recessive nonsyndromic deafness genes: a review. Front Biosci (Landmark Ed) 17: 2213-2236. [Crossref]
- Nance WE (2003) The genetics of deafness. Ment Retard Dev Disabil Res Rev 9: 109-119. [Crossref]
- Hilgert N, Smith RJ, Van Camp G (2009) Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat Res 681: 189-196. [Crossref]
- Wu CC, Liu TC, Wang SH, Hsu CJ, Wu CM (2011) Genetic characteristics in children with cochlear implants and the corresponding auditory performance. Laryngoscope 121: 1287-1293. [Crossref]
- Miyagawa M, Nishio SY, Hattori M, Moteki H, Kobayashi Y, et al. (2015) Mutations in the MYO15A gene are a significant cause of nonsyndromic hearing loss: massively parallel DNA sequencing-based analysis. Ann Otol Rhinol Laryngol 124: S158-S168. [Crossref]
- Liang Y, Wang A, Belyantseva IA, Anderson DW, Probst FJ, et al. (1999) Characterization of the human and mouse unconventional myosin XV genes responsible for hereditary deafness DFNB3 and shaker 2. Genomics61: 243-258. [Crossref]
- Belyantseva IA, Boger ET, Friedman TB (2003) Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc Natl Acad Sci U S A100: 13958-13963. [Crossref]
- Belyantseva IA, Boger ET, Naz S, Frolenkov GI, Sellers JR, et al. (2005) Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat Cell Biol 7: 148-156. [Crossref]
- Probst FJ, Fridell RA, Raphael Y, Saunders TL, Wang A, et al. (1998) Correction of deafness in shaker-2 mice by an unconventional myosin in a BAC transgene. Science 280: 1444-1447. [Crossref]
- Beyer LA, Odeh H, Probst FJ, Lambert EH, Dolan DF, et al. (2000) Hair cells in the inner ear of the pirouette and shaker 2 mutant mice. J Neurocytol 29: 227-240. [Crossref]
- Rehman AU, Bird JE, Faridi R, Shahzad M, Shah S, et al. (2016) Mutational Spectrum of MYO15A and the Molecular Mechanisms of DFNB3 Human Deafness. Hum Mutat 37: 991-1003. [Crossref]
- Nal N, Ahmed ZM, Erkal E, Alper OM, Luleci G, et al. (2007) Mutational spectrum of MYO15A: the large N-terminal extension of myosin XVA is required for hearing. Hum Mutat 28: 1014-1019. [Crossref]
- Belguith H, Aifa-Hmani M, Dhouib H, Said MB, Mosrati MA, et al. (2009) Screening of the DFNB3 locus: identification of three novel mutations of MYO15A associated with hearing loss and further suggestion for two distinctive genes on this locus. Genet Test Mol Biomarkers 13: 147-151. [Crossref]
- Cengiz FB, Duman D, Sirmaci A, Tokgoz-Yilmaz S, Erbek S, et al. (2010) Recurrent and private MYO15A mutations are associated with deafness in the Turkish population. Genet Test Mol Biomarkers 14: 543-550. [Crossref]
- Fattahi Z, Shearer AE, Babanejad M, Bazazzadegan N, Almadani SN, et al. (2012) Screening for MYO15A gene mutations in autosomal recessive nonsyndromic, GJB2 negative Iranian deaf population. Am J Med Genet A 158: 1857-1864. [Crossref]
- Woo HM, Park HJ, Baek JI, Park MH, Kim UK, et al. (2013) Whole-exome sequencing identifies MYO15A mutations as a cause of autosomal recessive nonsyndromic hearing loss in Korean families. BMC Med Genet 14: 72. [Crossref]
- Gao X, Zhu QY, Song YS, Wang GJ, Yuan YY, et al. (2013) Novel compound heterozygous mutations in the MYO15A gene in autosomal recessive hearing loss identified by whole-exome sequencing. J Transl Med 11: 284. [Crossref]
- Manzoli GN, Bademci G, Acosta AX, Felix TM, Cengiz FB, et al. (2016) Targeted Resequencing of Deafness Genes Reveals a Founder MYO15A Variant in Northeastern Brazil. Ann Hum Genet 80: 327-331. [Crossref]
- Mardis ER (2008) The impact of next-generation sequencing technology on genetics. Trends Genet 24: 133-41. [Crossref]
- Metzker ML (2010) Sequencing technologies - the next generation. Nat Rev Genet 11: 31-46. [Crossref]
- Shendure J, Ji H (2008) Next-generation DNA sequencing. Nat Biotechnol 26: 1135-1145.
- Wei X, Ju X, Yi X, Zhu Q, Qu N, et al. (2011) Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing. PloS one 6: e29500. [Crossref]
- Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11: 361-362. [Crossref]
- Yang T, Wei X, Chai Y, Li L, Wu H (2013) Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet J Rare Dis 8: 85. [Crossref]
- Kalay E, Uzumcu A, Krieger E, Caylan R, Uyguner O, et al. (2007) MYO15A (DFNB3) mutations in Turkish hearing loss families and functional modeling of a novel motor domain mutation. Am J Med Genet A 143: 2382-2389. [Crossref]
- Lezirovitz K, Pardono E, de Mello Auricchio MT, de Carvalho ESFL, Lopes JJ, et al. (2008) Unexpected genetic heterogeneity in a large consanguineous Brazilian pedigree presenting deafness. Eur J Hum Genet 16: 89-96. [Crossref]
- Shearer AE, Hildebrand MS, Webster JA, Kahrizi K, Meyer NC, et al. (2009) Mutations in the first MyTH4 domain of MYO15A are a common cause of DFNB3 hearing loss. Laryngoscope 119: 727-733. [Crossref]
- Anderson DW, Probst FJ, Belyantseva IA, Fridell RA, Beyer L, et al. (2000) The motor and tail regions of myosin XV are critical for normal structure and function of auditory and vestibular hair cells. Hum Mol Genet 9: 1729-1738. [Crossref]
- Manor U, Disanza A, Grati M, Andrade L, Lin H, et al. (2011) Regulation of stereocilia length by myosin XVa and whirlin depends on the actin-regulatory protein Eps8. Curr Biol 21: 167-172. [Crossref]
- Weber KL, Sokac AM, Berg JS, Cheney RE, Bement WM (2004) A microtubule-binding myosin required for nuclear anchoring and spindle assembly. Nature 431: 325-329. [Crossref]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, et al. (2015) RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347: 1254806. [Crossref]