TIP60 is multifunctional acetyltransferase highly expressed in heart cells, displays diverse functions including in regulation of transcription, autophagy, DNA damage response, cell cycle and stem cell function. Coincidentally, all of these cell activity participants in the maintenance of cardiovascular cell and the anti-cardiovascular disease. The conflictions exit between myocardial ischemic conditioning induce cardioprotection and the conditioning methods fail to be effective in humans, suggesting a strong demand of target with multipath effect to the cardiovascular disease (CVD). There are multiple association between the molecular pathway of TIP60 and the pathogenesis of CVD, but the correlation study between TIP60 and CVD is scant. Hence, we review the common action pathways for TIP60 and CD, to better understand the role of TIP60 in CVD.
TIP60, cardiovascular disease, autophagy, DNA damage response, stem cell
The acetyltranferase TIP60 (HIV-1 Tat interactive protein, 60 kD) is a member of the MYST family of histone acetyltransferases, and the expressed intensity of TIP60 in adult tissues is followed as: testis > heart > brain > kidney > liver > lung [1]. TIP60 is multifunctional acetyltransferase displays diverse functions in various cellular processes, including gene expression [2,3], autophagy [4], DNA damage response (DDR) [3,5], cell cycle, chromatin structure alternation, genomic instability, and stem cell maintenance and differentiation [6]. Interestingly, the autophagy maintains intracellular homeostasis of cardiovascular cells [7] and induces accumulated DNA damage, defective response to DSBs, increased chromosomal breakages and cell-cycle checkpoint defects in cardiovascular disease (CVD) and (or) models, suggesting a potential connection between the TIP60 function and the occurrence of CVD.
CVD is the leading global cause of death, creating an favourable environment for DNA repair of the damaged myocardium or enhancing the heart’s/myocardium’s tolerance to ischemia-reperfusion injury are the two new approaches for treating CVD [8]. Large amount of experimental research has demonstrated that all forms of myocardial ischemic conditioning can induce potent and effective cardioprotection [9-12], while none of these conditioning methods have been effective in humans:
a. Remote ischemic preconditioning did not improve clinical outcomes in 1,612 patients undergoing elective on-pump coronary artery bypass grafting [13].
b. Upper-limb remote ischemic preconditioning also did not exhibit any significant benefit in 1,385 patients undergoing elective cardiac surgery [14]. These ineffective results may be due to the underlying risk factors that interfere with cardiac conditioning and the effects of cardioprotective agents, suggesting future research should focus more on comorbid animal models and the finding of multiple pathways and target points [8].
At present, few studies center on the correlations of TIP60 with the development of cardiovascular cell (13 related articles, 5 directly related literature), and that less attention has been given to the TIP60 in CVD (5 related articles, no directly one). Herein, we summarize the effects and molecular pathway of TIP60 which may be associated with the development of cardiovascular cell and pathogenesis of CVD, suggesting a new and potent effective target of CVD.
Tip60 inducts autophagy and autophagy modulates cardiovascular cell homeostasis
Autophagy is a highly regulated process involving the removal of damaged proteins and organelles from cells and tissues through a lysosomal-mediated pathway. Earlier study uncovered the GSK3-TIP60-ULK1 autophagy-activating pathway linked with the growth factor deprivation to autophagy, demonstrating that activated GSK3 catalyzes phosphorylation of TIP60 at Ser86, which depends on a prior phosphorylation at Ser90 and results in higher affinity of TIP60 for ULK1 and increased acetylation and kinase activity of ULK1 [15]. In line with an essential role of TIP60 for autophagy induction is the previous observation that TIP60-/- mouse blastocysts fail to undergo implantation and die around embryonic day 3.5, when autophagy activity is strongly elicited for normal implantation [16].
Similarly, Sato, et al. first raised the GSK-3 isoforms may drive the differentiation of stem cell [17], and latter studies showed that the GSK-3 promotes embryonic stem cell differentiation into a cardiomyocyte lineage via Wnt/β-Catenin-independent mechanisms [18,19]. Moreover, Yu W, et al. suggested that the inactivation of the mTOR/ULK1-dependent pathway may serve as a key mechanism for the cardiac protective effect [20]. These results suggesting these mutual molecules and pathways in the TIP60 function and cardiovascular cell activity.
Accumulating evidence suggests that autophagy is necessary to maintain redox homeostasis, stress stimulate an increase in mitochondrial ROS to increase autophagic flux. Similarly, the induction of autophagy leads to a diminishment of oxidative stress, either through a change in metabolism or through the removal of ROS-producing organelles (e.g. mitochondria or peroxisomes) [21]. Autophagy contributes to the maintenance of intracellular homeostasis in most cells of cardiovascular origin, including cardiomyocytes, endothelial cells, and arterial smooth muscle cells [7]. Genetic defects in autophagy have been shown to exacerbate the propensity of laboratory animals to spontaneously develop cardiodegenerative disorders (such as myocardial infarction, cardiomyopathy and atherosclerosis) [22-26]. Moreover, pharmacological or genetic maneuvers that alter the autophagic flux have been shown to influence disease outcome in rodent models of several cardiovascular conditions, such as myocardial infarction, various types of cardiomyopathy and atherosclerosis [27-29].
The regulation mechanisms of Tip60 in DDR and DDR participants in anti-cardiovascular disease
DNA damage can be induced by several exogenous and endogenous stimulate factors, accumulated DNA damage causes persistent activation of the DNA damage response (DDR), genome instability and telomere attrition, and then repair factors are recruited to response the consequences of the DNA damage response (DDR) [30]. Double-strand break (DSB) as an important type of DNA damage are induced by exposure to ionizing radiation, which can be repaired by homologous recombination (HR) and non-homologous end-joining (NHEJ). Tip60 regulates the repair of DNA damage and plays a key role in DSB repair [31], the regulation mechanism can be summarized as:
Response to DDR: activation of ATM: The ATM protein kinase is a master regulator of the cell’s response to DNA damage through the phosphorylation of proteins involved in DNA repair and cell-cycle checkpoints [32]. Exposed to ionizing radiation induced DSB, and activated Tip60’s acetyltransferase activity, which leading to the acetylation of the ATM kinase and regulating the DDR [32]. These actions including chromodomain at the N-terminal of Tip60 and histone H3 trimethylated on lysine 9 induced the Tip60's acetyltransferase activity and the interaction between activated Tip60 and ATM is mediated by FATC domain which acetylates lysine 3016 of ATM [33,34].
Chromatin structure-Acetylation and ubiquitination of H2AX: H2AX is a histone variant that differs from H2A. When the DSBs have been induced, the MRN complex (MRE11, RAD50, and NBS1) binds to broken DNA ends and recruits active ATM, ATR, and/or DNA protein kinase, which further leading to the initial phosphorylation of H2AX (γ-H2AX) [35-37]. The phosphorylated H2AX participants in the accumulation of DNA repair factors such as the MRN complex, 53BP1, and BRCA1 [38,39]. Moreover, once DSB happens, TIP60 regulates the acetylation-dependent ubiquitination of H2AX and leads to the release of H2AX from damaged chromatin through the forming the TIP60-UBC13 complex, which further promotes enhanced histone dynamics and stimulates the DDR [40].
Genome stability:
NuA4/TIP60 complex-DSB repair is a particularly harmful DNA damage, which leads to loss of genetic material and genomic instability. The NuA4/TIP60 complex plays a key role in DSB repair [41], which could rapidly acetylate H4, H2A and H2AX and facilitating the relaxation of the chromatin at the DSB [41-43]. Meanwhile, both the TIP60 and NuA4 directly interact with γ-H2AX, which further facilitate chromatin dynamics in DNA repair [44, 45].
TIP60 controls 53BP1-dependent DNA repair-53BP1 is a bivalent chromatin reader, and is directly ubiquitylated by RNF168, leading to 53BP1 oligomerization, which is critical for its relocalization to DSBs [46]. Strikingly, both the TIP60 and 53BP1 are binding to H2AK15ub and H4K20me2, H4K20 methylation is rapidly recognized by the double Tudor domain of 53BP1 at DSB, while the activities of deacetylases and de-ubiquitinases of H2AK15 are important in shifting the balance between TIP60 and 53BP1 [47,48]. TIP60 shares common nucleosomal histone targets with 53BP1 on nucleosomes and create an acetylation/ubiquitylation switch at H2AK15 to regulates DSB repair.
DNA damage and these consequences of the DDR (eg, cellular apoptosis and senescence) are present in atherosclerotic plaques, ataxia-telangiectasia, Werner Syndrome and Hutchinson-Gilford Progeria Syndrome, pulmonary arterial hypertension, myocardial Infarction and heart failure, myocarditis and rheumatic heart disease. Herein, the role and mechanisms of DNA Damage in CVD have been summarized as:
Atherosclerotic Plaques-Human samples and animal models’ studies found that atherosclerotic plaques contain accumulated DNA damage and activated DNA damage response elements. Compared with normal tissue, human atherosclerotic plaques have more DSBs and activation of ATM [49], oxidative DNA damage is also increased [50]. Increased apoptosis and senescence have also been shown in the cellular components of atherosclerotic lesions [51-53].
Ataxia-Telangiectasia-Ataxia-telangiectasia is an autosomal recessive disorder [54], the heterozygotes of the ataxia-telangiectasia mutated allele have an increased death risk from ischemic heart disease compared with noncarriers [55]. The cells from Ataxia-Telangiectasia patients also display a defective response to DSBs, increased chromosomal breakages, and cell-cycle checkpoint defects.
Werner Syndrome and Hutchinson-Gilford Progeria Syndrome: Werner syndrome (WS) is also a rare autosomal recessive disease, is characterized by premature onset of the signs of aging [56] and myocardial infarction, stroke and malignancies are the major causes of death in patients with WS. WS mutations lead to truncation or instability of the WRN protein [56,57], which shows single-strand annealing activity [58,59]. Cells from WS patients are hypersensitive to genotoxic reagents and display various chromosomal abnormalities, and WRN knock-down cells also exhibit chromosomal abnormalities and increased amounts of DSB and enhanced DDR [60]. These observations suggest that WRN is involved in DNA replication and repair, telomere maintenance, and gene transcription [61]. Hutchinson-Gilford Progeria Syndrome (HGPS) is a rare but severe premature aging syndrome, HGPS cells or tissue samples also display increased sensitivity to genotoxic reagents, activated ATM and their downstream kinases, elevated Foci of 53BP1 and amount of γH2AX, and increased DNA damage and defects in DDR [62-67].
Pulmonary Arterial Hypertension: Pulmonary arterial hypertension (PAH) is associated with oxidative stress and sustained inflammation which are known to increase DNA damage [68]. Compared with healthy tissues/cells, higher levels of DNA damage and makers (53BP1 and γ-H2AX) have been detected in PAH lungs and remodeled arteries as well as in animal models of pulmonary hypertension [68,69]. The loss of breast cancer 2 protein (BMPR2) usually damaged the DNA damage repair, and the down-regulation of BMPR2 in PAH pulmonary arterial endothelial cells (PAECs) increased susceptibility to DNA damages [70]. Topoisomerase DNA II binding protein 1 (TopBP1) is also involved in PAH susceptibility [71], which is important in maintaining genome integrity by preventing DNA damage during replication [72-74], further strengthens the fact that impaired DNA repair is involved in PAH susceptibility.
Myocardial Infarction and Heart Failure: A multifarious types of DNA damage including single-strand break (SSB), double-strand break (DSB) and DNA oxidation have been detected in the heart of infarcted zone, and the activation of DDR strongly influences cardiac remodeling after myocardial infarction [75-77]. Similarly, the activated DDR has also been observed in cardiomyocytes of the heart failure mice induced by pressure-overload [78] as well as the end-stage heart failure patients with [79]. Higo T et al. demonstrated that in the cardiomyocytes of heart failure mice induced by pressure-overload, the unrepaired DNA SSB is accumulated, which further activates continuing DDR and causes gene expression of inflammatory cytokines through NF-kB signaling [80].
In short, the accumulation of DNA damage activates persistent DDR, genome instability, and telomere attrition, further lead to progressive cellular senescence and apoptosis presenting in CVD. Moreover, persistent DDR promotes the secretion of proinflammatory cytokines, chemokines and growth factors in cardiovascular cells [81], which may accelerate atherosclerosis and/or increase plaque vulnerability [82,83], also participate in the pathology of the myocarditis [84] and rheumatic heart disease [85]. Tip60 regulates the repair of DNA damage and plays a key role in DSB repair [31] through the activation of ATM and modulating the acetylation and ubiquitination of H2AX, controlling the formation of NuA4/TIP60 complex and 53BP1-dependent DNA repair, which may participant in the treatment of CVD.
Tip60 ensures the normal cell-cycle in cardiovascular cell: As described, DSB can be repaired by HR and NHEJ. Interestingly, these functions of HR and NHEJ normally act through the cell cycle to modify the broken DNA [86,87]. The ATM protein kinase is a master regulator of the cell’s response to DNA damage through the phosphorylation of proteins involved in DNA repair and cell-cycle checkpoints [88]. DeRan M, et al. demonstrated that Tip60 and transformation/transactivation domain-associated protein (TRRAP) are associated with histone gene promoters at the G1/S-phase boundary, histone H4 acetylation at histone gene promoters increases at the G1/S-phase transition [89]. Mo F, et al. identified CDK1-cyclin B phosphorylated Ser90 of TIP60, which elicited TIP60-dependent acetylation of Aurora B at Lys215 protected the phosphorylation of its activation loop, and promoted accurate chromosome segregation in mitosis [90]. Niida H, et al. found that Active reductase (RNR) subunits RRM1 bounds to a damage site in a Tip60-dependent manner, while lacking Tip60 binding RRM1 mutants failed to rescue an impaired DNA repair in RRM1-depleted G1-phase cells [91]. Taubert S, et al. declared that ectopically expressed E2F1 bounding to unrelated HAT Tip60, induced recruitment of five subunits of the Tip60 complex (Tip60, TRRAP, p400, Tip48, and Tip49) to target promoters to chromatin occurred in late G1 following serum stimulation [92]. In conclusion, TIP60 mediates the progression of the cell cycle by maintaining the genome integrity and facilitating the G1/S phase transition during the G1 and S phase, ensuring the faithful chromatin segregation during the M phase.
In the developing mouse heart, DNA synthesis, karyokinesis and cytokinesis of cardiomyocyte become arrested by the third postnatal week [93]. Recent evidence indicates that adult cardiomyocytes can re-enter the cell cycle and divide it [94-96]. Tip60 is highly expressed in the developing heart [97,98], while Tip60-heterozygous mice do not have a haploinsufficient phenotype at adult stages and that Tip60-null mice die at the blastocyst stage [99]. In adult Tip60-heterozygous B-lymphocytes, imposition of Myc-induced stress was recently shown to induce lymphomagenesis, revealing a haploinsufficient phenotype accompanied by diminished cell-cycle control [100]. Moreover, stress induced hypertrophy in Tip60+/- adult hearts, and caused increased activity in cardiomyocyte cell-cycle, which is accompanied by reduced apoptosis [101]. All of these results indicate that the role of Tip60 in maintaining cell-cycle inhibition and promoting apoptosis in the adult myocardium.
GSK3/p-Tip60 promote and maintain the differentiation of cardiomyocytes: Stem cells are a kind of multipotent cells with self-renewing ability. Under certain conditions, it can be differentiated into multiple functional cells. Stem cell is an under differentiated, immature cell with the potential to regenerate a variety of tissues and organs and human body function, the medical community known as "universal cells". stem cells can differentiate into Endothelial cell, while cardiovascular health relies on a fully functional endothelium. Now days, more and more Clinical studies have reported that safety, efficacy and dosage response to stem cell populations [102]. In vitro, studies in recent years have also implicated GSK-3 (glycogen synthase kinase-3) is a vital regulator of embryonic stem cells differentiation [103-105]. Under certain condition, GSK-3β, isoform of GSK-3, playing a central role in regulating differentiation into the cardiomyocyte lineage [108-110], deletion of GSK-3β is sufficient to sustain the proliferative capacity of stem cells.
Tip60 can hold the maintenance and renewal of stem cells. It was found that the SANT domain of P400 and the HAT domain of Tip60 bind to each other, which locate in embryonic stem cells, neural stem cells, and hematopoietic stem and progenitor cells, and inhibit the activity of Tip60 acetyltransferase [111], Tip60-p400 genes promote stem cells to undergo self-renewal, and to switch off the genes that transform stem cells into other kinds of cells [112], but the molecular mechanisms responsible for these effects have Not yet been identified. Experimental studies suggested that Tip60 is required for pluripotency and represses the downstream processes of differentiation [113,114] in embryonic stem cells.
Currently, increasing studies have revealed that Tip60 also induce the DNA damage response, apoptosis, and cell-cycle regulation of cardiomyocytes [115,116]. Just as in pluripotent cells of the blastocyst [8], Tip60 is indispensable for maintaining cytoactive of differentiated cardiomyocytes. In addition, some other experiments further show that Tip60 modulates expression of serum response factor-dependent cardiac genes [117]. Through phosphorylating TIP60-Ser86, GSK-3 activates acetyltransferase TIP60, then directly acetylates and stimulates the protein kinase ULK1, which is required for autophagy [118,119]. On the one hand, default of GSK-3β enhance the proliferative capacity of stem cells, which differentiate into Endothelial cells; On the other hand, GSK-3 can promote acetyltransferase TIP60, which keep the viability of differentiated cardiomyocytes. Thus, Stem cells, TIP60 and cardiovascular disease interact with each other via certain biomolecules.
TIP60 is highly expressed in heart cells, which participant in the regulation of autophagy, DDR, cell cycle and stem cell differential. The activated GSK3 catalyzes phosphorylation of TIP60 at Ser86, results in higher affinity of TIP60 for ULK1 and increased acetylation and kinase activity of ULK1, which further activate autophagy. The induction of autophagy leads to a diminishment of oxidative stress and contributes to the maintenance of intracellular homeostasis in most cardiovascular cells. Genetic defects in autophagy or alteration of autophagic flux have been shown to exacerbate the propensity of laboratory animals to spontaneously develop cardiodegenerative disorders. Moreover, the GSK-3 also β promotes embryonic stem cell differentiation into a cardiomyocyte lineage via Wnt/β-Catenin-independent mechanisms, and the inactivation of the mTOR/ULK1-dependent pathway may also serve as a key mechanism for the cardiac protective effect. These results suggesting that the TIP60 may participant in the forming and maintaining of cardiomyocyte function through modulation of the autophagic flux, and the dysfunction of TIP60 may damage the cardiomyocyte and induce CVD.
Exposed to ionizing radiation induces DSB, activates Tip60’s acetyltransferase activity, leading to the acetylation of the ATM kinase which further induce the initial phosphorylation of H2AX (γ-H2AX) and regulating the DDR. TIP60 regulates the acetylation-dependent ubiquitination of H2AX and leads to the release of H2AX from damaged chromatin through the forming the TIP60-UBC13 complex, which further promotes enhanced histone dynamics and stimulates the DDR. Moreover, TIP60 forms the NuA4/TIP60 that interact with γ-H2AX facilitating chromatin dynamics in DNA repair, and controls 53BP1-dependent DNA repair. TIP60 also mediates the progression of the cell cycle by maintaining the genome integrity and facilitating the G1/S phase transition during the G1 and S phase, ensuring the faithful chromatin segregation during the M phase to play against DNA damage. The accumulation of DNA damage activates persistent DDR, genome instability, and telomere attrition, further lead to progressive cellular senescence and apoptosis presenting in CVD including atherosclerotic plaques, ataxia-telangiectasia, Werner Syndrome etc. While, Tip60 facilitates chromatin dynamics and regulates the repair of DNA damage, which participant in the anti-cardiovascular disease. Herein, the roles and pathway mechanisms of TIP60 in modulation of cardiomyocyte and its effect of anti-cardiovascular disease through regulating autophagy and DDR, suggesting TIP60 acts a new and potent effective target of CVD. Herein, we summarized the action mechanisms of TIP60 in the activation of autophagy and DDR, suggesting a potential anti-cardiovascular disease role of TIP60 (Figure 1).
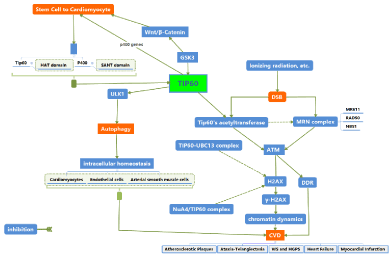
Figure 1. 1) The GSK3-TIP60-ULK1 autophagy-activating pathway linked with the growth factor deprivation to autophagy, which may be necessary to maintain the intracellular homeostasis of cardiovascular cells. 2) Exposed to ionizing radiation induces DSB, activates Tip60’s acetyltransferase activity, leading to the acetylation of the ATM kinase and regulating the DDR. TIP60 forms the NuA4/TIP60 that interact with γ-H2AX facilitating chromatin dynamics in DNA repair, and controls 53BP1-dependent DNA repair. 3) TIP60 promotes and maintain the differentiation of cardiomyocytes. 4) All of these pathways of TIP60 may act important roles in the pathophysiology and treatment of CVD.
This study was supported by the Natural Science Foundation of Liaoning province of China (2015020672).
The research was conducted in the absence of any commercial or financial relationships that could be misconstrued as a potential conflict of interest. We confirm that all authors contributed to this manuscript and have approved the final article.
- Meloche J, Pflieger A, Vaillancourt M, Paulin R, Potus F, et al. (2014) Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 129: 786-797. [Crossref]
- Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, et al. (2004) Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 306: 2084-2087. [Crossref]
- Squatrito M, Gorrini C, Amati B (2006) Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends cell Biol 16: 433-442. [Crossref]
- Lin SY, Li TY, Liu Q, Zhang C, Li X, et al. (2012) GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 336: 477-481. [Crossref]
- Mo F, Zhuang X, Liu X, Yao PY, Qin B, et al. Acetylation of Aurora B by TIP60 ensures accurate chromosomal segregation. Nat Chem Biol 12: 226-232. [Crossref]
- Yamada HY (2012) Human Tip60 (NuA4) complex and cancer. Colorectal cancer biology-from genes to tumor. InTech.
- Bravo-San Pedro JM, Kroemer G, Galluzzi L (2017) Autophagy and Mitophagy in Cardiovascular Disease. Circ Res 120: 1812-1824. [Crossref]
- Terashvili M, Bosnjak ZJ (2018) Stem Cell Therapies in Cardiovascular Disease. J Cardiothorac Vasc Anesth 1-14.
- Heusch G, Botker HE, Przyklenk K, Redington A, Yellon D (2015) Remote ischemic conditioning. J Am Coll Cardiol 65: 177-195. [Crossref]
- Heusch G (2013) Cardioprotection: Chances and challenges of its translation to the clinic. Lancet 381: 166-175. [Crossref]
- Schwartz Longacre L, Kloner RA, Arai AE, Baines CP, Bolli R, et al. (2011) New horizons in cardioprotection recommendations from the 2010 National Heart, Lung, and Blood Institute Workshop. Circulation 124: 1172-1179. [Crossref]
- Heusch G (2015) Molecular basis of cardioprotection signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res 116: 674-699. [Crossref]
- Hausenloy DJ, Candilio L, Evans R, Ariti C, Jenkins DP, et al. (2015) Remote ischemic preconditioning and outcomes of cardiac surgery. New Engl J Med 373: 1408-1417. [Crossref]
- Meybohm P, Kohlhaas M, Stoppe C, Gruenewald M, Renner J, et al. (2018) RIPHeart [Remote Ischemic Preconditioning for Heart Surgery) Study: Myocardial dysfunction, postoperative neurocognitive dysfunction, and 1-year follow-up. J Am Heart Assoc 7: 7. [Crossref]
- Lin SY, Li TY, Liu Q, Zhang C, Li X, et al. (2018) GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 336: 477-481. [Crossref]
- Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, et al. (2008) Autophagy is essential for preimplantation development of mouse embryos. Science 321: 117-120. [Crossref]
- Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH (2004) Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med 10: 55-63. [Crossref]
- Kerkela R, Kockeritz L, MaCauly K, Zhou J, Doble BW, et al. (2008) Deletion of GSK-3β in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest 118: 3609-3618. [Crossref]
- Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR (2007) Functional Redundancy of GSK-3αand GSK-3βin Wnt/β-Catenin Signaling Shown by Using an Allelic Series of Embryonic Stem Cell Lines. Dev Cell 12: 957-971. [Crossref]
- Yu W, Zha W, Ren J (2018) Exendin-4 and Liraglutide Attenuate Glucose Toxicity-Induced Cardiac Injury through mTOR/ULK1-Dependent Autophagy. Oxid Med Cell Longev 5396806.
- Yan Y, Finkel T (2017) Autophagy as a regulator of cardiovascular redox homeostasis. Free Radic Biol Med 109:108-113. [Crossref]
- Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, et al. (2013) Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest 123: 5284-5297. [Crossref]
- Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, et al. (2008) Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci USA 105: 9745–9750. [Crossref]
- Liu X, Ye B, Miller S, Yuan H, Zhang H, et al. (2012) Ablation of ALCAT1 mitigates hypertrophic cardiomy- opathy through effects on oxidative stress and mitophagy. Mol Cell Biol 32: 4493–4504. [Crossref]
- McLendon PM, Ferguson BS, Osinska H, Bhuiyan MS, James J, et al. (2014) Tubulin hyperacetylation is adaptive in cardiac proteotoxicity by promoting autophagy. Proc Natl Acad Sci USA 111: E5178–E5186. [Crossref]
- Finckenberg P, Eriksson O, Baumann M, Merasto S, Lalowski MM, et al. (2012) Caloric restriction ameliorates angiotensin II-induced mitochondrial remodeling and cardiac hypertrophy. Hypertension 59: 76-84. [Crossref]
- Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G (2017) Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov 16: 487-511. [Crossref]
- Green DR, Levine B (2014) To be or not to be? How selective autophagy and cell death govern cell fate. Cell 157: 65-75. [Crossref]
- Fuchs Y, Steller H (2015) Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol 16: 329-344. [Crossref]
- Menezo Y, Dale B, Cohen M (2010) DNA damage and repair in human oocytes and embryos: a review. Zygote 18: 357-365. [Crossref]
- Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, et al. (2002) Acetylation of histone H4by Esa1 is required for DNA double-strand break repair. Nature 419: 411-415. [Crossref]
- Sun Y, Jiang X, Chen S, Fernandes N, Price BD (2005) A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A 102: 13182-13187. [Crossref]
- Jiang X, Sun Y, Chen S, Roy K, Price BD (2006) The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J Biol Chem 281: 15741–15746. [Crossref]
- Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, et al. (2009) Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol 11: 1376-1382. [Crossref]
- Petrini JH, Stracker TH (2003) The cellular response to DNA double-strand breaks: Defining the sensors and mediators. Trends Cell Biol 13: 458-462. [Crossref]
- Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, et al. (2002) Histone H2A variants H2AX and H2AZ. Curr Opin Genet Dev 12: 162-169. [Crossref]
- Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, et al. (2007) DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol 27: 7028-7040. [Crossref]
- Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, et al. (2003) Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 5: 675-679. [Crossref]
- Tsukuda T, Fleming AB, Nickoloff JA, Osley MA (2005) Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature 438: 379-383. [Crossref]
- Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, et al. (2007) DNA Damage-Dependent Acetylation and Ubiquitination of H2AX Enhances Chromatin Dynamics. Mol Cell Biol 27: 7028-7040. [Crossref]
- Rossetto D, Truman AW, Kron SJ, Côté J (2010) Epigenetic modifications in double-strand break DNA damage signaling and repair. Clin Cancer Res 16: 4543-4552. [Crossref]
- Courilleau C, Chailleux C, Jauneau A, Grimal F, Briois S, et al. (2012) The chromatin remodeler p400 ATPase facilitates Rad51-mediated repair of DNA double-strand breaks. J Cell Biol 199: 1067-1081. [Crossref]
- Soria G, Polo SE, Almouzni G (2012) Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell 46: 722-734. [Crossref]
- van Attikum H, Fritsch O, Gasser SM (2007) Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J 26: 4113-4125. [Crossref]
- van Attikum H, Fritsch O, Hohn B, Gasser SM (2004) Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 119: 777-788. [Crossref]
- Lottersberger F, Bothmer A, Robbiani DF, Nussenzweig MC, de Lange T (2013) Role of 53BP1 oligomerization in regulating double-strand break repair. Proc Natl Acad Sci USA 110: 2146-2151. [Crossref]
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, et al. (2006) Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127: 1361-1373. [Crossref]
- Jacquet K, Fradet-Turcotte A, Avvakumov N, Lambert JP, Roques C, et al. (2016) The TIP60 complex regulates bivalent chromatin recognition by 53BP1 through direct H4K20me binding and H2AK15 acetylation. Mol Cell 62: 409-421. [Crossref]
- Mahmoudi M, Gorenne I, Mercer J, Figg N, Littlewood T, et al. (2008) Statins use a novel Nijmegen breakage syndrome-1-dependent pathway to accelerate DNA repair in vascular smooth muscle cells. Circ Res 103: 717-725. [Crossref]
- Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM (2002) Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 106: 927-932. [Crossref]
- Minamino T, Yoshida T, Tateno K, Miyauchi H, Zou Y, et al. (2003) Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation 108: 2264-2269. [Crossref]
- Bennett MR, Evan GI, Schwartz SM (1995) Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J Clin Invest 95: 2266-2274. [Crossref]
- Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, et al. (2002) Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 105: 1541-1544. [Crossref]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, et al. (1995) A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268: 1749-1753. [Crossref]
- Su Y, Swift M (2000) Mortality rates among carriers of ataxia-telangiectasia mutant alleles. Ann Intern Med 133: 770-778. [Crossref]
- Huang S, Lee L, Hanson NB, Lenaerts C, Hoehn H, et al. (2006) The spectrum of WRN mutations in Werner syndrome patients. Hum Mutat 27: 558-567. [Crossref]
- Moser MJ, Oshima J, Monnat RJ Jr (1999) WRN mutations in Werner syndrome. Hum Mutat 13: 271-279. [Crossref]
- Kudlow BA, Kennedy BK, Monnat RJ Jr (2007) Werner and HutchinsonGilford progeria syndromes: Mechanistic basis of human progeroid diseases. Nat Rev Mol Cell Biol 8: 394-404. [Crossref]
- Rossi ML, Ghosh AK, Bohr VA (2010) Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst) 9: 331-344. [Crossref]
- Galvan N, Lim S, Zmugg S, Smith MT, Zhang L (2008) Depletion of WRN enhances DNA damage in HeLa cells exposed to the benzene metabolite, hydroquinone. Mutat Res 649: 54-61. [Crossref]
- Singh DK, Ahn B, Bohr VA (2009) Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging. Biogerontology 10: 235-252. [Crossref]
- Varela I, Cadinanos J, Pendas AM, Gutiérrez-Fernández A, Folgueras AR, et al. (2005) Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 437: 564-568. [Crossref]
- Schreiber KH, Kennedy BK (2013) When lamins go bad: Nuclear structure and disease. Cell 152: 1365-1375. [Crossref]
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, et al. (2005) Genomic instability in laminopathy-based premature aging. Nat Med 11: 780-785. [Crossref]
- Liu Y, Rusinol A, Sinensky M, Wang Y, Zou Y (2006) DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A. J Cell Sci 119: 4644-4649. [Crossref]
- Liu Y, Wang Y, Rusinol AE, Sinensky MS, Liu J, et al. (2008) Involvement of xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A. FASEB J 22: 603-611. [Crossref]
- Liu GH, Barkho BZ, Ruiz S, Diep D, Qu J, et al. (2011) Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 472: 221-225. [Crossref]
- Meloche J, Pflieger A, Vaillancourt M, Paulin R, Potus F, et al. (2014) Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 129: 786-797. [Crossref]
- Ranchoux B, Meloche J, Paulin R, Boucherat O, Provencher S, et al. (2016) DNA damage and pulmonary hypertension. Int J Mol Sci 17. [Crossref]
- Li M, Vattulainen S, Aho J, Orcholski M, Rojas V, et al. (2014) Loss of bone morphogenetic protein receptor 2 is associated with abnormal DNA Repair in pulmonary arterial hypertension. Am J Respir Cell Mol Biol 50: 1118-1128. [Crossref]
- De Jesus Perez VA, Yuan K, Lyuksyutova MA, Dewey F, Orcholski ME, et al. (2014) Whole-exome sequencing reveals TopBP1 as a novel gene in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 189: 1260-1272. [Crossref]
- Lee Y, Katyal S, Downing SM, Zhao J, Russell HR, et al. (2012) Neurogenesis requires TopBP1 to prevent catastrophic replicative DNA damage in early progenitors. Nat Neurosci 15: 819-826.
- Yamane K, Wu X, Chen J (2002) A DNA damage-regulated BRCT-containing protein, TopBP1, is required for cell survival. Mol Cell Biol 22: 555-566. [Crossref]
- Mäkiniemi M, Hillukkala T, Tuusa J, Reini K, Vaara M, et al. (2001) BRCT domain-containing protein TopBP1 functions in DNA replication and damage response. J Biol Chem 276: 30399-30406. [Crossref]
- Shukla PC, Singh KK, Quan A, Al-Omran M, Teoh H, et al. (2011) BRCA1 is an essential regulator of heart function and survival following myocardial infarction. Nat Commun 2: 511-593.
- Zingarelli B, Cuzzocrea S, Zsengeller Z, Salzman AL, Szabó C (1997) Protection against myocardial ischemia and reperfusion injury by 3-aminobenzamide, an inhibitor of poly (ADP-ribose) synthetase. Cardiovasc Res 36: 205-215. [Crossref]
- Cordis GA, Maulik G, Bagchi D, Riedel W, Das DK (1998) Detection of oxidative DNA damage to ischemic reperfused rat hearts by 8-hydroxydeoxyguanosine formation. J Mol Cell Cardiol 30: 1939-1944. [Crossref]
- Raymond AR, Norton GR, Sareli P, Woodiwiss AJ, Brooksbank RL (2013) Relationship between average leucocyte telomere length and the presence or severity of idiopathic dilated cardiomyopathy in black Africans. Eur J Heart Fail 15: 54-60. [Crossref]
- Mourkioti F, Kustan J, Kraft P, Day JW, Zhao MM, et al. (2013) Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nat Cell Biol 15: 895-904. [Crossref]
- Higo T, Naito AT, Sumida T, Shibamoto M, Okada K, et al. (2017) DNA single-strand break-induced DNA damage response causes heart failure. Nat Commun 8: 15104. [Crossref]
- Campisi J (2013) Aging, cellular senescence, and cancer. Annu Rev Physiol 75: 685-705. [Crossref]
- Manabe I (2011) Chronic inflammation links cardiovascular, metabolic and renal diseases. Circ J 75: 2739-2748. [Crossref]
- Simon DI (2012) Inflammation and vascular injury: Basic discovery to drug development. Circ J 76: 1811-1818. [Crossref]
- Abdel-Wahab BA, Metwally ME, El-khawanki MM, Hashim AM (2014) Protective effect of captopril against clozapine-induced myocarditis in rats: role of oxidative stress, proinflammatory cytokines and DNA damage. Chem Biol Interact 216: 43-52. [Crossref]
- Rastogi M, Sarkar S, Makol A, Singh RS, Saikia UN, et al. (2018) Anti-endothelial cell antibody rich sera from rheumatic heart disease patients induces proinflammatory phenotype and methylation alteration in endothelial cells. Genes Dis 5: 275-289.
- Arnaudeau C, Lundin C, Helleday T (2001) DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol 307: 1235-1245. [Crossref]
- Lieber MR (2008) The mechanism of human nonhomologous DNA end joining. J Biol Chem 283: 1-5. [Crossref]
- Sun Y, Jiang X, Chen S, Fernandes N, Price BD (2005) A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A 102: 13182-13187. [Crossref]
- DeRan M, Pulvino M, Greene E, Zhao J (2008) Transcriptional activation of histone genes requires NPAT-dependent recruitment of TRRAP-Tip60 complex to histone promoters during the G1/S phase transition. Mol Cell Biol 28: 435-447. [Crossref]
- Mo F, Zhuang X, Liu X, Yao PY, Qin B, et al. (2016) Acetylation of aurora B by TIP60 ensures accurate chromosomal segregation. Nat Chem Biol 12: 226-232. [Crossref]
- Niida H, Katsuno Y, Sengoku M, Shimada M, Yukawa M, et al. (2010) Essential role of Tip60-dependent recruitment of ribonucleotide reductase at DNA damage sites in DNA repair during G1 phase. Genes Dev 24: 333-338. [Crossref]
- Taubert S, Gorrini C, Frank SR, Parisi T, Fuchs M, et al. (2004) E2F-dependent histone acetylation and recruitment of the Tip60 acetyltransferase complex to chromatin in late G1. Mol Cell Biol 24: 4546-4556. [Crossref]
- Soonpaa MH, Field LJ (1998) Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res 83: 15-26. [Crossref]
- Engel FB, Schebesta M, Duong MT, Lu G, Ren S, et al. (2005) p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev 19: 1175-1187. [Crossref]
- Kuhn B, del Monte F, Hajjar RJ, Chang YS, Lebeche D, et al. (2007) Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat Med 13: 962-969. [Crossref]
- Bersell K, Arab S, Haring B, Kühn B (2009) Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138: 257-270. [Crossref]
- Kim MS, Merlo X, Wilson C, Lough J (2006) Co-activation of atrial natriuretic factor promoter by Tip60 and serum response factor. J Biol Chem 281:15082-15089. [Crossref]
- Lough JW (2002) Transient expression of TIP60 protein during early chick heart development. Dev Dyn 223: 419-425. [Crossref]
- Hu Y, Fisher JB, Koprowski S, McAllister D, Kim MS, et al. (2009) Homozygous disruption of the Tip60 gene causes early embryonic lethality. Dev Dyn 238: 2912-2921. [Crossref]
- Gorrini C, Squatrito M, Luise C, Syed N, Perna D, et al. (2007) Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 448: 1063-1067. [Crossref]
- Fisher JB, Kim MS, Blinka S, Ge ZD, Wan T, et al. (2012) Stress-Induced Cell-Cycle Activation in Tip60 Haploinsufficient Adult Cardiomyocytes. PLoS One 7 :e31569. [Crossref]
- Reed DM, Foldes G, Harding SE, Mitchell JA (2013) Stem cell-derived endothelial cells for cardiovascular disease: A therapeutic perspective. Br J Clin Pharmacol 75: 897-906. [Crossref]
- Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, et al. (2005) LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 132: 885-896. [Crossref]
- Chou YF, Chen HH, Eijpe M, Yabuuchi A, Chenoweth JG, et al. (2008) The growth factor environment defines distinct pluripotent ground states in novel blastocyst-derived stem cells. Cell 135: 449-461. [Crossref]
- Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH (2004) Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med 10: 55-63. [Crossref]
- Silva J, Smith A (2008) Capturing pluripotency. Cell 132: 532-536. [Crossref]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, et al. (2008) The ground state of embryonic stem cell self-renewal. Nature 453: 519-23. [Crossref]
- Kerkela R, Kockeritz L, Macaulay K, Zhou J, Doble BW, et al. (2008) Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest 118: 3609-3618. [Crossref]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, et al. (2008) The ground state of embryonic stem cell self-renewal. Nature 453: 519-523. [Crossref]
- Doble BW1, Patel S, Wood GA, Kockeritz LK, Woodgett JR (2007) Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell 12: 957-971. [Crossref]
- Lin SY, Li TY, Liu Q, Zhang C, Li X, et al. (2012) GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 336: 477-481. [Crossref]
- Samuelson AV, Narita M, Chan HM, Jin J, de Stanchina E, et al. (2005) p400 is required for E1A to promote apoptosis. J Biol Chem 280: 21915-21923. [Crossref]
- Chen PB, Hung JH, Hickman TL, Coles AH, Carey JF, et al. (2013) Hdac6 regulates Tip60-p400 function in stem cells. Elife 2: e01557. [Crossref]
- Yang Y, Han X, Guan J, Li X (2014) Regulation and function of histone acetyltransferase MOF. Front Med 8: 79-83. [Crossref]
- Fazzio TG, Huff JT, Panning B (2008) An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell 134: 162-174. [Crossref]
- Fisher JB, Kim MS, Blinka S, Ge ZD, Wan T, et al. (2012) Stress-induced cell-cycle activation in Tip60 haploinsufficient adult cardiomyocytes. PLoS One 7: e31569. [Crossref]
- Gogna R, Madan E, Khan M, Pati U, Kuppusamy P (2013) P53's choice of myocardial death or survival: Oxygen protects infarct myocardium by recruiting p53 on NOS3 promoter through regulation of p53-Lys(118) acetylation. EMBO Mol Med 5: 1662-1683. [Crossref]
- Hu Y, Fisher JB, Koprowski S, McAllister D, Kim MS, et al. Homozygous disruption of the Tip60 gene causes early embryonic lethality. Dev Dyn 238: 2912-2921. [Crossref]
- Kim MS, Merlo X, Wilson C, Lough J (2006) Co-activation of Atrial Natriuretic Factor Promoter by Tip60 and Serum Response Factor. J Biol Chem 281: 15082-15089. [Crossref]