Introduction
A 55 year old man with past medical history of hypothyroidism presented to the emergency department complaining of 3 months history of progressively worsening abdominal distention, scrotal swelling, lower extremity edema and NYHA III dyspnea. On further questioning, the patient reported having 3-4 incidences of sudden falls with no syncope which occurred when attempting to stand from a sitting position. Besides his history of hypothyroidism, patient had no significant past medical history, he reported compliance with his thyroid replacement therapy. On social history he denied any history of tobacco, alcohol or illicit drug use. He was not familiar with any medical history for his family. He was a very active long distance runner and swimmer and had spent 5 years in active duty in the military before leaving it to work in the construction business. During that he denied any chemical, or biological exposure.
Physical exam and diagnostic studies
The patient had a blood pressure of 143/100 mm Hg, heart rate of 103 bpm, respiratory rate of 18/min and had an oxygen saturation of 97% on room air. He was noted to have bilateral lower extremity pitting edema which extended midway up the abdomen associated with ascites, genital inspection showed scrotal edema. Initial labs were significant for Hemoglobin of 21, hematocrit of 64, Brain Natriuretic Peptide (BNP) of 18000, albumin level of 1.3, dyslipidemia with low density lipoprotein of 309, Urine analysis showed frothy urine with high urine protein on dipstick, urine protein to creatinine ratio was 11.1. Random urine protein was 1 gm per deciliter. Contrast Tomography (CT) abdomen was remarkable for large volume ascites along with hepatomegaly with hepatic steatosis.
Hospital course
Patient was admitted for further evaluation of nephrotic syndrome, and was initially started on intravenous furosemide 40 mg twice daily. Human influenza virus, hepatitis panel, rapid plasma reagin antinuclear antibody testing and JAK2V617 mutation was negative; Complements C3 and C4 were normal. Erythropoeitin (EPO) was 56.8. Renal ultrasound demonstrated no gross structural abnormalities. Serum protein electrophoresis and immunotyping demonstrated a polyclonal distribution of IgG, IgA, and IgM with unremarkable kappa/lambda ratio and restricted heterogeneity all consistent with a non-selective proteinuria.
EKG showed sinus rhythm with left axis deviation low voltage QRS complexes and poor R wave progression (Figure 1). A 2 D Echocardiogram obtained on day 3 showed small left ventricular (LV) cavity, with thickened left and right ventricular walls (Movie Clip 1 and 2), systolic anterior motion of the anterior leaflet of the mitral valve (SAM) (Figure 2), enlarged septum with septum measuring 31 mm with LV outflow max gradient of 34 mm Hg , mean gradient of 19 mm Hg and velocity of 295 cm/sec, normal left ventricular wall motion with speckling and increased echogenicity , estimated LV ejection fraction (LVEF ) of 65-70%, abnormal relaxation pattern, normal sized right and left atria and a small pericardial effusion (Figure 3). LV Strain imaging showed Global Longitudinal strain of -7.47% (normal -18%) (Figure 4) with regional strain values greatly reduced in the basal and mid LV, yet preserved at the LV apex.
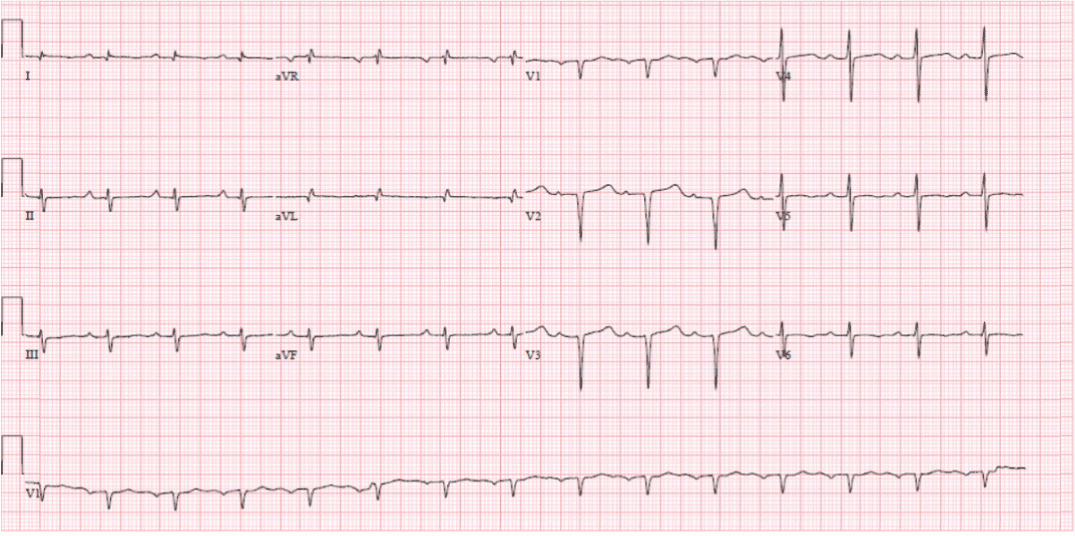
Figure 1. EKG showed sinus rhythm with left axis deviation low voltage QRS complexes and poor R wave progression.
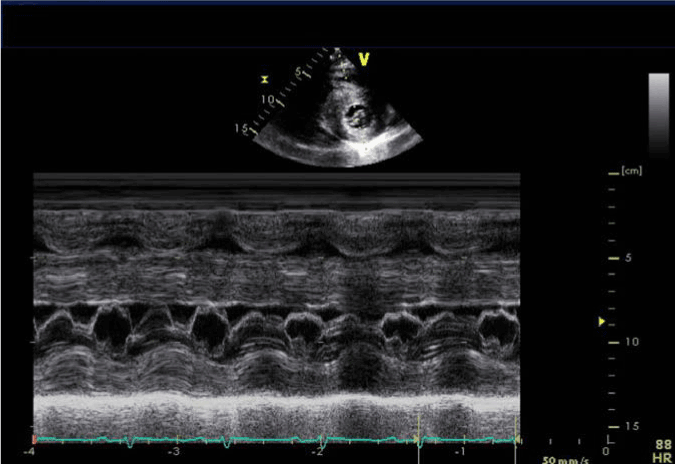
Figure 2. Parasternal short axis (PSAX) with M- mode echocardiography showing systolic anterior motion of the anterior leaflet of the mitral valve.
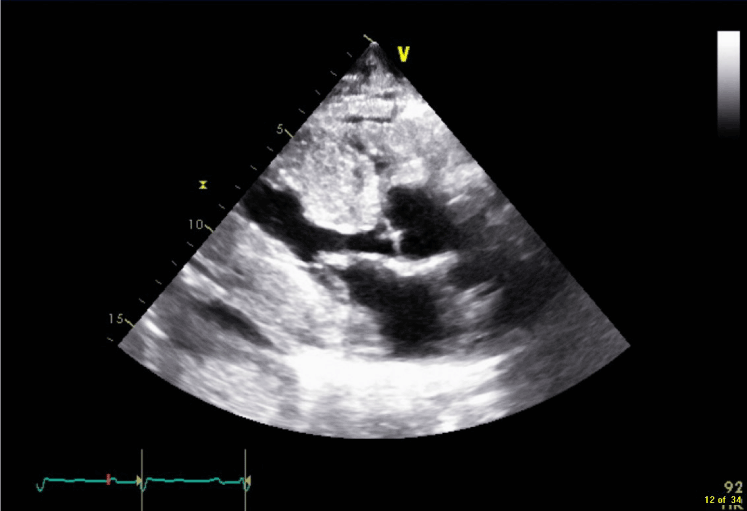
Figure 3. 2 D Echo Parasternal long axis (PLAX) view showing LV hypertrophy with enlarged septum and SAM of the anterior leaflet of the mitral valve which is thickened. A small pericardial effusion is also seen.
The patient underwent a kidney biopsy which revealed lambda light chain restricted AL-amyloidosis., under light microscopy there was deposition of eosinophilic amorphous amyloid material in the glomeruli, interstitium and walls of arterioles. In the immunofluorescence study the amyloid materials was only positive for lambda light chain; and in the electron microscopic evaluation, numerous thin and non-branching amyloid fibrils were identified. These amyloid fibrils were randomly distributed and measured 7 to 10 nm in length.
As the echo findings suggested Amyloid heart disease with HOCM physiology, he underwent a right heart catheterization (RHC) and endomyocardial biopsy. RHC showed normal Pulmonary Capillary Wedge Pressure of 4 cc of Hg, normal mean PA pressure of 11 millimeters of Hg and normal pulmonary vascular resistance of 2.25 Woods units. It demonstrated a reduced cardiac output of 3.11 liters/minute measured by thermodilution method, there was no evidence of intra-cardiac shunts by oximetry.
Endomyocardial biopsy confirmed extensive accumulation of lambda light chain restricted AL-amyloidosis in the cardiac parenchyma with no evidence of any other type of amyloid (Figure 5 and 6). In addition to accumulation of large amounts of amyloid-like amorphous material in the intercellular stroma and vascular walls, amyloid materials were also extending into or totally replacing cardiomyocytes. Under Electron Microscopy, the amyloid fibrils were seen to have penetrated cell membranes and replaced myofibrils (Figure 7 and 8).
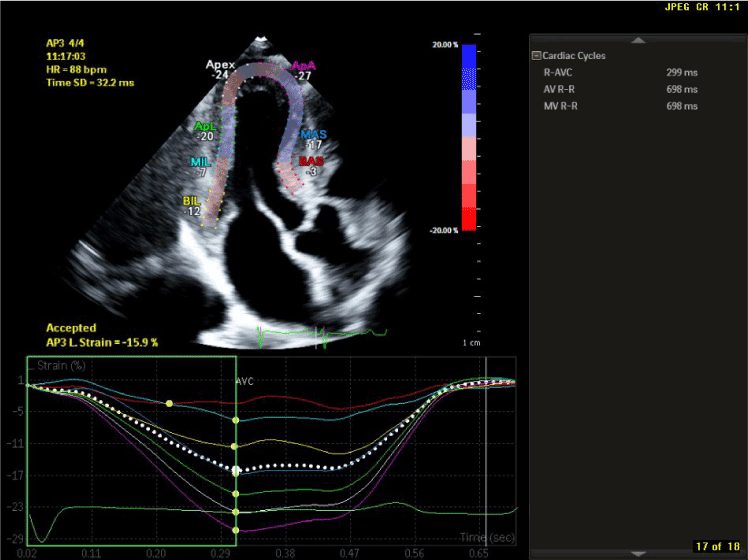
Figure 4. LV Strain imaging in 3 chamber Apical long axis view showing regional strain values greatly reduced in the basal and mid LV, yet preserved at the LV apex.
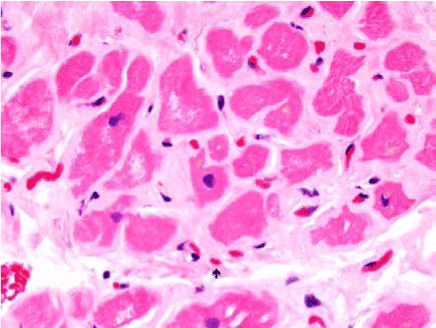
Figure 5. Hematoxylin – Eosin (H & E) Staining: Myocardial fibers (dark pink) surrounded by amorphous amyloid fibrils (light pink). Amyloid is also seen to have infiltrated the walls of the blood vessels. (Arrow)
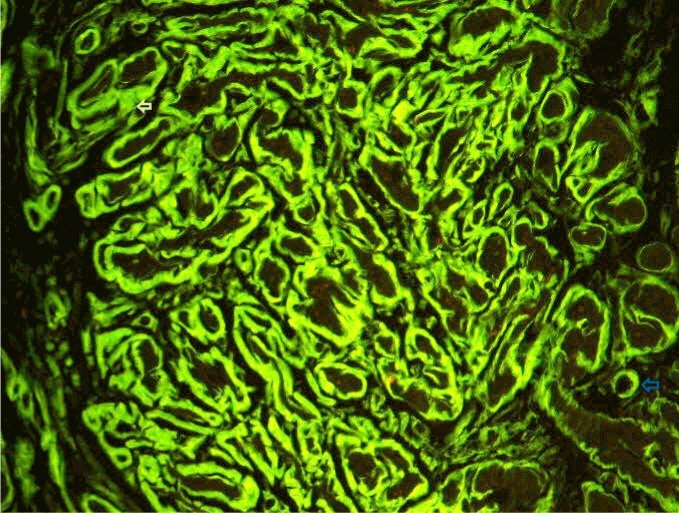
Figure 6. Immunofluorescence showing 2 to 3+ reaction for lambda light chain in the perimuscular stroma (white arrow) and the small arteries (blue arrow).

Figure 7. Electron microscopy at 1500 x magnification showing deposition and infiltration of amyloid fibrils (thick black arrowhead) replacing myocytes along with injured mitochondria and apoplectic nuclei (thin black arrows).
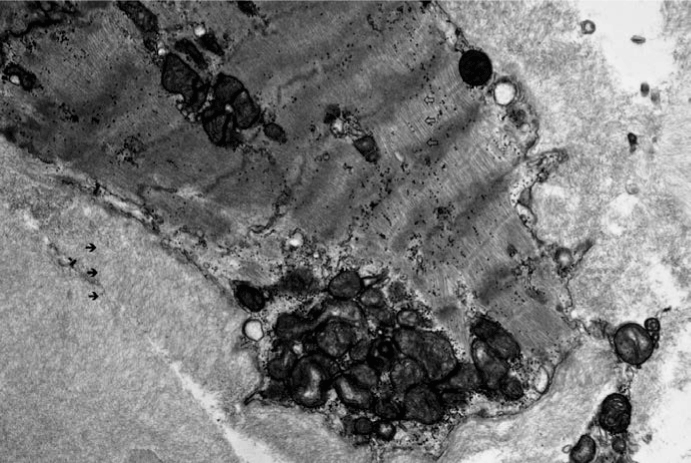
Figure 8. Electron microscopy at 4000x magnification showing deposition and infiltration of amyloid fibrils in the interstitial space (thick black arrows) replacing myocytes along with injured mitochondria and apoplectic nuclei. Small area of normal appearing myocytes seen (block arrows)
Bone marrow biopsy demonstrated amyloid depositions with plasmacytosis, lambda light chain restriction, and normal cytogenetics. Urine protein electrophoresis showed lambda light chain clone, a urine M component is 0.7%, and a kappa to lambda ratio of 0.1.
The patient met all four of the international myeloma working group criteria for AL amyloidosis with cardiac involvement [1]. He was initiated on metoprolol 12.5 mg twice daily and combination of Cyclophosphamide, bortezomib and dexamethasone for plasma cell dyscrasia with follow-up in hematology-oncology clinic. A 30 day monitor placed for complaints of lightheadedness after 6 weeks of follow-up revealed premature atrial complexes. A repeat echo at 6 month of follow-up showed improvement complete resolution of the systolic anterior motion of the anterior leaflet of the mitral valve (Figure 9) (Movie Clip 3). There was concentric left ventricular hypertrophy, however LV septum thickness was reduced to 24 mm with LVOT maximum gradient at 9 mm and mean gradient at 5 mm with peak velocity of 151 cm/s enlarged left and right atria. Strain imaging showed a basal LS of -8.5% and relative apical sparing similar to the previous echocardiogram with 2 dimensional speckle tracking longitudinal strain bulls eye plot demonstrating the “ cherry on top ” finding characteristic of cardiac amyloidosis (Figure 10).
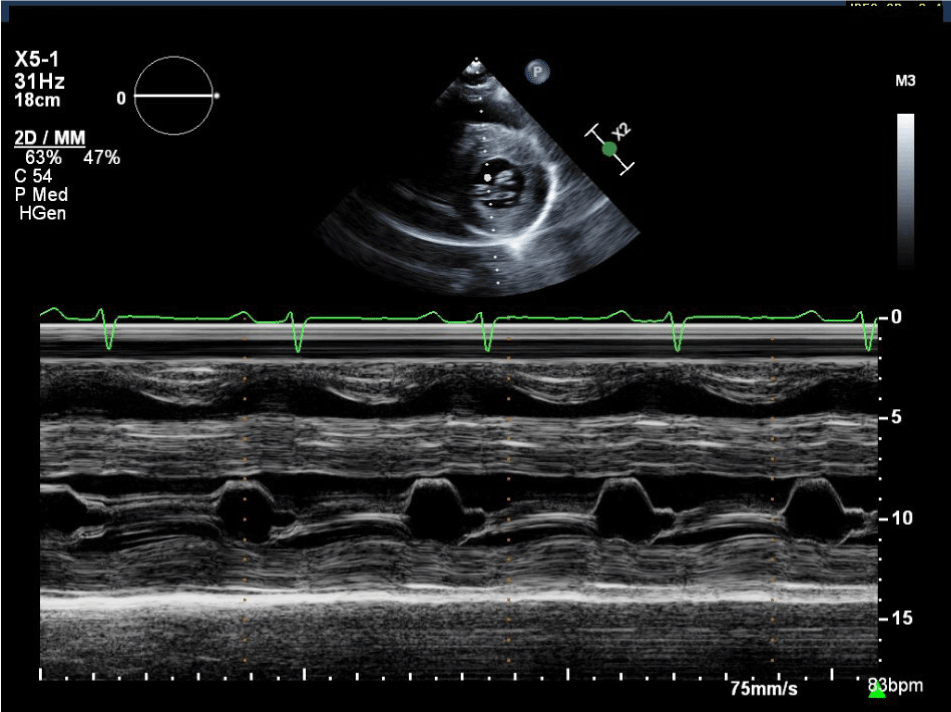
Figure 9. Parasternal short axis (PSAX) with M- mode echocardiography showing resolution of systolic anterior motion of the anterior leaflet of the anterior leaflet of the mitral valve.
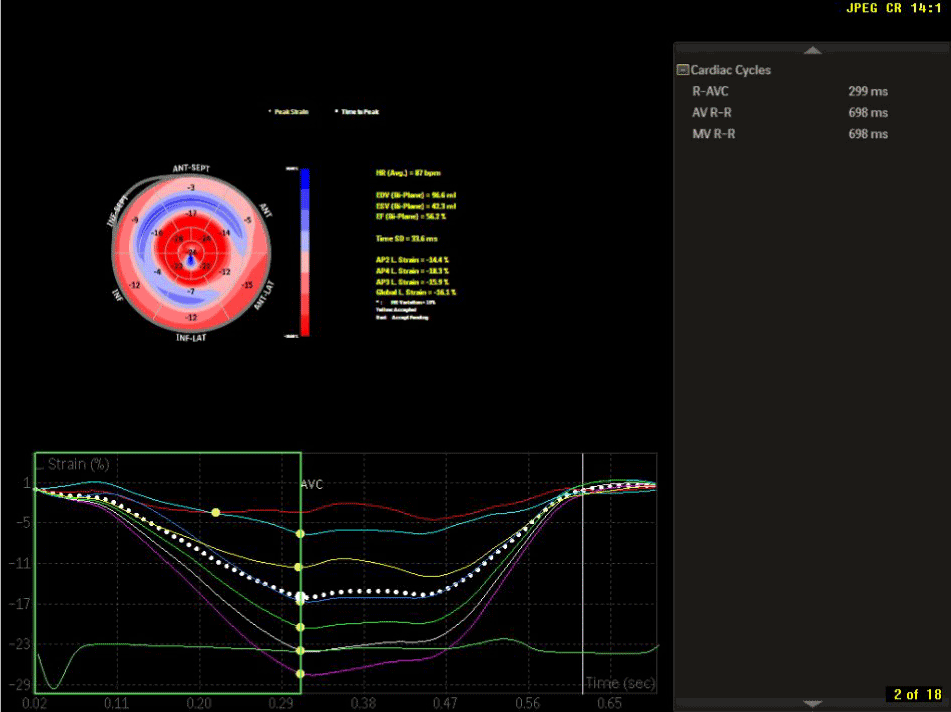
Figure 10. Echocardiography strain imaging with “cherry on top “finding characteristic for cardiac amyloid.
Discussion
This case is significant for a rare presentation of AL cardiac amyloidosis with echocardiographic signs of Left Ventricular Outflow Tract obstruction similar to a Hypertrophic Obstructive Cardiomyopathy. We believe that our patient had normal sized atria at diagnosis due to early diagnosis and treatment. He had resolution of his outflow obstruction after therapy with significant reduction in Left ventricular septal thickness. Individual cases of similar presentation have been previously reported [2,3], and a case series estimated a 4% incidence of cases of cardiac amyloidosis that present with outflow obstruction [4].
Identifying cardiac amyloid from HOCM is essential as HOCM has a better prognosis compared to amyloidosis with cardiac involvement in which death is seen in less than a year in the absence of therapy. Our patient had characteristic echocardiographic signs of HOCM namely outflow tract gradient and Systolic anterior motion of the mitral valve while EKG, non-cardiac involvement and speckled appearance of his myocardium suggested cardiac amyloidosis.
However cases with outflow obstruction may present with syncopal events or exertional dyspnea. Age of onset is usually above 50 years. Low voltage waves in the limb and the precordial leads similar to our patient’s EKG are the most common ECG finding in cardiac amyloidosis and are present in over 50% of patients with cardiac amyloid. A normal ECG may be present in 15% of hypertrophic cardiomyopathy patients, usually those with localized hypertrophy. Atrial arrhythmias are common along with conduction system abnormalities due to amyloid deposition [5].
Echocardiogram reveals a normal to small LV cavity size, increased biventricular wall thickness while Doppler imaging shows elevated LV filling pressures which is characteristic and severely impaired longitudinal LV function with a normal LV ejection fraction shows an enhanced speckled pattern with biventricular enlargement which is characteristic but not specific to amyloidosis. In one series this finding was present in 26% of patients [5].
In less than 5% of cases with cardiac amyloid, asymmetric septal thickening may be present with a LV outflow tract gradient mimicking HOCM. Syncopal events in such cases can be explained due to orthostatic hypotension due to reduced cardiac output due to LVOT obstruction, diastolic dysfunction, or autonomic nervous system dysfunction due to amyloidosis [3]. A specific pattern of longitudinal strain characterized by worse longitudinal strain in the mid and basal ventricle with relative sparing of the apex may help distinguish LV infiltration because of amyloid from true ventricular hypertrophy of hypertensive heart disease or hypertrophic cardiomyopathy. Among patients with AL amyloidosis, the mean basal strain, a measure of longitudinal LV function, was a powerful predictor of clinical outcome and was superior to standard 2-dimensional echocardiographic, Doppler flow measurements, and simple tissue velocity indexes [6-8]. A separate study in 2012 showed that a 2D LV global longitudinal strain of < -11.78% was shown to be an independent predictor of survival [9].
This necessitates caution and possibly avoidance of diuretic therapy as these patients are pre load dependent. Echocardiography can also reveal left atrial dysfunction including abnormal left atrial strain imaging. A study in 2005 showed lower left atrial systolic strain and reduced atrial global strain in 60% of patients with cardiac amyloidosis [10].
Cardiac magnetic resonance (CMR) imaging is a useful diagnostic and prognostic tool. Late gadolinium enhancement (LGE) on CMR identified cardiac involvement in 47% of patients with known systemic amyloidosis and normal wall thickness on echocardiogram. Both LV and RV LGE are seen while diffuse atrial LGE is characteristic in cardiac amyloidosis. Sub endocardial T1 mapping is shortened in cardiac amyloidosis, and T1 mapping may identify cardiac involvement at an earlier stage when compared with overt LGE images [11]. CMR has demonstrated increased extracellular volume in the hearts of patients with biopsy-proven amyloidosis, even in the absence of (LGE), which suggests that absence of LGE does not rule out early cardiac amyloidosis [12].
Radionuclide imaging of cardiac amyloidosis can be performed currently using single-photon emission computed tomography where direct amyloid imaging agents (I-123–labeled serum amyloid P component), bone imaging agents (Tc-99m pyrophosphate or Tc-99m 3, 3-diphosphono-1, 2-propanodicarboxylic acid [DPD]), and agents to image cardiac sympathetic innervation (I-123 metaiodobenzylguanidine) are available for use [12]. In 1 study, Tc-99m DPD was able to differentiate between AL and ATTR amyloidosis noninvasively, with patients with all ATTR showing DPD uptake and none of the patients with AL showing DPD uptake. [13].
Definitive diagnosis in patients with evidence of a monoclonal gammopathy who are suspected of having cardiac AL amyloidosis remains a tissue biopsy showing amyloid deposits. An endomyocardial biopsy is done at some centers and is sometimes preferred as it gives the advantage of hemodynamic assessment during the procedure. Sections are stained with H and E stain which shows amyloid fibrils interspersed between normal fibrils. Immunohistochemistry shows infiltration of amyloid fibrils into myocytes and arterial and arteriolar walls. Electron microscopy is significant for destruction of normal myocyte structure with sheets of amyloid fibrils, apoplectic nuclei and mitochondria. On the other hand EM images of HOCM show hypertrophied cardiomyocytes with bizarre forms with Y shaped branching with frequent side to side junction or having a whorled appearance around a fibrous core.
Treatment of cardiac amyloidosis is twofold – a) Treatment of underlying plasma cell dyscrasia and b) treatment of heart failure. Treating heart failure in AL associated cardiac amyloidosis with diuretics is the mainstay of therapy. ACE inhibitors are poorly tolerated due to hypotension, calcium channel blockers are contraindicated while beta blockers may not improve outcomes. This makes cases of cardiac amyloid with outflow obstruction challenging as these patients are intravascularly dry in spite of significant right sided failure. AL Amyloidosis has seen an improvement in prognosis with combination of bortezomib and dexamethasone however prognosis is still poor with onset of heart failure. Early treatment of underlying plasma cell dyscrasia may improve heart failure in some patients. Disopyramide has been used successfully to decrease the LV outflow gradient in a case report [14]. Beta blockers have been used with caution. Cardiac transplantation in AL amyloidosis has been performed, but the outcome depends on extremely careful selection of patients. One-year overall survival was 64% after LV assist device (LVAD) implantation, with no difference between amyloidosis and non-amyloidosis patients, but with a very poor survival if the LV end-diastolic dimension was <46 mm [15]. A 2015 study of 16 patients with systemic amyloidosis who were treated with a combination of a amyloid depleting compound and a Ig – Anti serum amyloid P component antibody showed early promise in reducing amyloid burden and improving liver and renal function [15-17].
Author contributions
2021 Copyright OAT. All rights reserv
Drs Abdulbaki, Apte and Douglas were instrumental in drafting the article, analyzing the data and doing a review of current and past literature. Drs Garcia and Apte were responsible for obtaining the data for the case. Dr Lockhart and Dr Gu provided the histopathological images and interpretation for the case. Case approved by Dr Abdulbaki and reviewed by Dr Modi.
References
- International Myeloma Working Group: Criteria for Systemic AL amyloidosis.
- Wang J, Marzolf A, Zhang JC, Owens A, Han Y (2016) Cardiac Amyloidosis Masked as Hypertrophic Cardiomyopathy: A Case Report. Cardiol Res 7: 178-180. [Crossref]
- Boufidou A, Mantziari L, Paraskevaidis S, Karvounis H, Nenopoulou E ,et al. (2010) An Interesting Case of Cardiac Amyloidosis Initially Diagnosed as Hypertrophic Cardiomyopathy. Hellenic J Cardiol 51: 552-557. [Crossref]
- Dinwoodey DL, Skinner M, Maron MS, Davidoff R, Ruberg FL (2008) Light-chain amyloidosis with echocardiographic features of hypertrophic cardiomyopathy. Am J Cardiol 101: 674-676. [Crossref]
- Falk RH, Alexander KM, Liao R, Dorbala S (2016) AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J Am Coll Cardiol 68: 1323-1341. [Crossref]
- Falk RH, Plehn JF, Deering T, Schick EC Jr, Boinay P, et al. (1987) Sensitivity and specificity of the echocardiographic features of cardiac amyloidosis. Am J Cardiol 59: 418-422. [Crossref]
- Koyama J, Falk RH (2010) Prognostic significance of strain Doppler imaging in light-chain amyloidosis. JACC Cardiovasc Imaging 3:333–342. [Crossref]
- Koyama J, Ray-Sequin PA, Falk RH (2003) Longitudinal myocardial function assessed by tissue velocity, strain, and strain rate tissue Doppler echocardiography in patients with AL (primary) cardiac amyloidosis. Circulation 107: 2446–2452. [Crossref]
- Phelan D, Collier P, Thavendiranathan P, Popović ZB, Hanna M, et al. (2012) Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart 98: 1442–1448. [Crossref]
- Baccouche H, Maunz M, Beck T, Gaa E, Banzhaf M, et al. (2012) Differentiating cardiac amyloidosis and hypertrophic cardiomyopathy by use of three-dimensional speckle tracking echocardiography. Echocardiography 29: 668–677. [Crossref]
- Modesto KM, Dispenzieri A, Cauduro SA, Lacy M, Khandheria BK, et al. (2005) Left atrial myopathy in cardiac amyloidosis: implications of novel echocardiographic techniques. Eur Heart J 26:173–179. [Crossref]
- Maceira AM, Prasad SK, Hawkins PN, Roughton M, Pennell DJ (2008) Cardiovascular magnetic resonance and prognosis in cardiac amyloidosis. J Cardiovasc Magn Reson 10: 54. [Crossref]
- Falk RH, Quarta CC, Dorbala S (2014) How to image cardiac amyloidosis. Circ Cardiovasc Imaging 7: 552-562. [Crossref]
- Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, et al. (2005) Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol 46:1076–1084. [Crossref]
- Philippakis AA, Falk RH (2012) Cardiac Amyloidosis Mimicking Hypertrophic Cardiomyopathy with Obstruction: Treatment with Disopyramide. Circulation 125: 1821-1824. [Crossref]
- Falk RH, Alexander KM, Liao R, Dorbala S (2016) AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J Am Coll Cardiol 68: 1323-1341. [Crossref]
- Richards DB, Cookson LM, Berges AC, Barton SV, Lane T, et al. (2015) Therapeutic Clearance of Amyloid by Antibodies to Serum Amyloid P Component. N Engl J Med 373: 1106-1114. [Crossref]