Abstract
Diamond-Blackfan anemia (DBA) presenting in adulthood is uncommon and presents with diverse clinical manifestations, different from classical cases. The notion of DBA as a ribosomopathy has been clarified, but many features of it´s pathophysiology particularly role of sex hormones remain unknown. We report the occurrence of DBA in a young adult, with switch from mild to severe transfusion dependent bone marrow (BM) failure after sex-reassignment intervention.
Key words
Diamond-Blackfan anemia, sex-reassigment surgery, inherited bone marrow failure syndromes
Abbreviations
Hb: Hemoglobin; WBC: White Blood Cells; PTL: Platelets; HDE: High Dose Estrogens; Sx: Surgery; T: Testosterone
Introduction
Diamond-Blackfan anemia is an inherited bone marrow failure syndrome (IBMFS), characterized by macrocytic anemia with erythroblastopenia, presence of congenital abnormalities, and cancer predisposition, mostly diagnosed in the first year of life [1,2]. Recent data shows that approximately 45% of the cases are inherited in an autosomal dominant pattern, however, penetrance is reduced, for which a wide range of severity within a family can be found [3,4].
Although several theories regarding the physiopathology of DBA have been proposed, it is now widely categorized as a ribosomopathy. Mutations in distinct ribosomal protein (RP) genes have been encountered in more than 50% of the cases appearing to be the syndrome manifestations, the result of the RP haploinsufficiency [1,5,6].
Evidence supports that this defect in ribosome biogenesis confers an intrinsic disorder of erythropoiesis rather than the result of an immune-mediated red cell failure [2]. Differential expression levels of specific RPs limiting for ribosome assembling in certain tissues, diminished translation of mRNAs for anti-apoptotic factors, direct toxicity related to the accumulation of unassembled RPs or precursor complexes, and most recently, alteration of the telomere biology suggesting that DNA repair and ribosome biogenesis may be interconnected, are some of the postulations to explain the relation between ribosomal dysfunction and BM failure [7-9].
Once an accurate diagnosis is stablished supportive care with appropriate use of transfusions is the mainstay of therapy. Approximately 20% to 30% of the patients will recover spontaneously after initial supportive care, for the reminder a trial with a dose of 2mg/kg of prednisone, or the glucocorticoid equivalent, should be given with an expected response rate of 80% [1,2,10].
Hematopoietic stem cell transplantation continues to be the only curative treatment for the hematologic manifestations of DBA, being considered only for steroid-refractory and steroid-dependent (prednisone dose >0.3mg/kg/d) that are transfusion dependent [10,11]. Long-term survival with this intervention is achieved in 65% to 75% of the patients [12-14].
We hereby present a case of a patient with diagnosis of DBA, who developed transfusion dependency after sex-reassignment male-to-female surgery.
Case report
A 26-year-old woman with history of mild non-classical DBA, was seen in our clinic because of new onset of pallor, increasing feeling of fatigue and poor concentration.
Her initial diagnosis was made at the age of 19 years when being asymptomatic she was evaluated as a possible donor for her brother who had severe transfusion dependent classical DBA. The HLA-typing revealed that she was a match, but she was found to have mild macrocytic anemia with reticulocytopenia, elevated Hemoglobin F (HbF), elevated erythrocyte adenosine deaminase activity (ADA) and erythroid hypoplasia in the BM study. As hemoglobin (Hb) levels were stable and patient was symptomatic, no intervention with transfusion support or medical therapy was initiated.
Our patient is a transsexual who was started on high-dose estrogen (HDE) therapy (estradiol 3 mg daily) at the age of 21 years and underwent a sexual-reassignment male-to-female surgery 8 months prior to our assessment. Until the time of surgery with regards to her DBA, she remained asymptomatic and never required any transfusion. Laboratory investigations including testosterone levels, were within normal limits, except for a mild macrocytic anemia (Hb concentration 120 g/L) (Figure 1).
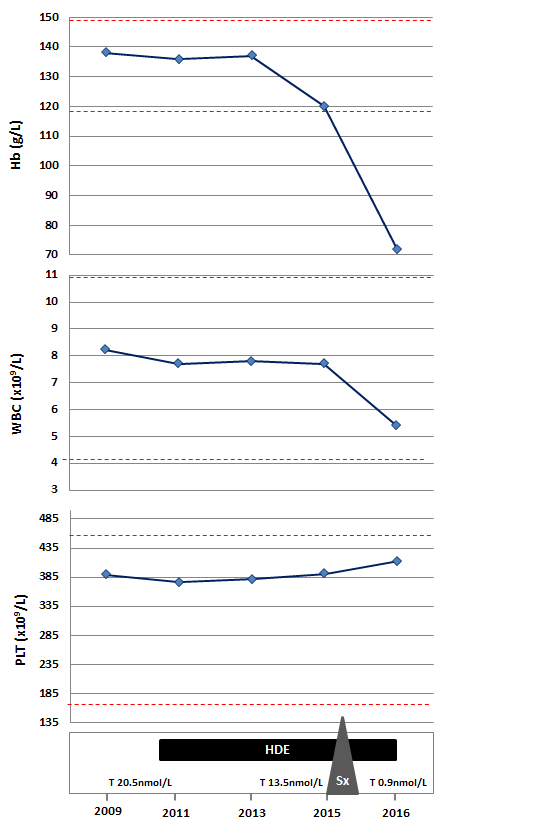
Figure 1. Serial hematologic and testosterone parameters in relation with interventions.
On the current visit besides her anemia related symptoms, there were no other remarkable findings on physical examination. However, her cell blood counts were notable for showing severe anemia with Hb concentration of 72 g/L (Figure 1). Further investigations to rule out other possible causes of anemia, including vitamin deficiencies, hypothyroidism, hemolysis and viral serologies were done, but the only parameter that was expectedly found to be abnormal was the testosterone level (0.9 nmol/L; range 8.0 – 32.0).
The patient started needing regular red blood cell transfusions. Relation of anemia with androgen deprivation and exposure to high dose estrogens, as well as possible therapeutic options were extensively discussed with the patient, but considering patient´s interests, only conservative treatment with transfusion support was started.
Disscusion
We describe a rare clinical case of DBA presenting in young adulthood, better defined as a non-classical DBA that illustrates clearly the phenotypic variability of the disease [1]. Diversity of hematologic manifestations and congenital abnormalities even in familial cases sharing an established DBA RP mutation, underscores the importance of a careful physical and hematologic evaluation of family members when a new case of DBA is identified [15].
In recent years several aspects of DBA pathophysiology have been uncovered leading to a wider understanding of its molecular basis. However, the growing identification of non-classical cases and the lack of clear correlation between genotype and phenotype, evidence the influence of the low penetrance of causing mutations and the potential effect of environmental factors and modifier genes [15,16].
In transsexual people, cross-sex hormone therapy is an important component of medical treatment. Initial administration of HDE that confers feminizing effects, is followed by an irreversible sex assignment surgery in which orchiectomy is performed [17]. In the case presented, we hypothesize that the withdrawal of androgen effects was the factor disrupting the equilibrium in compensated hematopoiesis inducing transfusion dependency and progression to severe DBA.
After 1948 when Hamilton initially described the physiological and metabolic changes that occur following orchiectomy, other researchers outlined that the decrease in hemoglobin concentration is directly correlated with testosterone levels [18]. The mechanism of the erythropoietic effects of androgens appears to be complex, with evidence for stimulation of erythropoietin secretion and direct effect on the BM. However, anemia related to hypogonadism has only been reported to be mild, with a median decrease in Hb concentration of 12 g/L [19,20].
On the other hand, the role of androgen therapy in some cases of BM failure syndromes with telomerase complex mutations has been well described. In vitro androgens demonstrated to have a role on stimulating telomerase gene expression in hematopoietic cells, including CD34+ cells and lymphocytes [21,22]. Recent findings by Townsley et al. in 27 patients with telomere diseases treated with danazol, regarding the relation between increased telomerase activity, modest telomere leukocyte elongation and improvement in peripheral blood counts, support this mechanism of action as the explanation for their efficacy in hematopoietic diseases [23,24].
Alter et al. were able to demonstrate that telomeres in DBA were shorter when compared with normal individual, suggesting that telomere biology might also be implicated in the pathophysiology [9]. Under this evidence we suggest that an environmental factor, such as testosterone imbalance might be the trigger for a change in clinical manifestations in a patient with stable mild DBA.
Conclusion
Advances in the understanding of DBA molecular basis have led to increase identification of non-classical DBA cases with distinct clinical features from classical cases. Despite this many features of the pathophysiology, including the impact of environmental factors remains unanswered. As in other types of IBMFS, the implication of endocrine dysfunction in DBA patients should be extensively reviewed, as there is increasing evidence that enhances the importance of sex hormone balance in the homeostasis of hematopoietic stem cells.
References
- Vlachos A, Ball S, Dahl N, Alter BP, Sheth S, et al. (2008) Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol 142: 859-876. [Crossref]
- Lipton JM, Ellis SR (2009) Diamond-Blackfan anemia: diagnosis, treatment, and molecular pathogenesis. Hematol Oncol Clin North Am 23: 261-282. [Crossref]
- Lipton JM, Atsidaftos E, Zyskind I, Vlachos A (2006) Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: an update from the Diamond Blackfan Anemia Registry. Pediatr Blood Cancer 46: 558-564. [Crossref]
- Orfali KA, Ohene-Abuakwa Y, Ball SE (2004) Diamond Blackfan anaemia in the UK: clinical and genetic heterogeneity. Br J Haematol 125: 243-252. [Crossref]
- Doherty L, Sheen MR, Vlachos A, Choesmel V, O'Donohue MF, et al. (2010) Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am J Hum Genet 86: 222-228. [Crossref]
- Chae H, Park J, Lee2021 Copyright OAT. All rights reservomal protein mutations in Korean patients with Diamond-Blackfan anemia. Exp Mol Med 46: e88. [Crossref]
- Shimamura A (2006) Inherited bone marrow failure syndromes: molecular features. Hematology Am Soc Hematol Educ Program. [Crossref]
- Ellis SR, Massey AT (2006) Diamond Blackfan anemia: A paradigm for a ribosome-based disease. Med Hypotheses 66: 643-648. [Crossref]
- Alter BP, Giri N, Savage SA, Rosenberg PS (2015) Telomere length in inherited bone marrow failure syndromes. Haematologica 100: 49-54. [Crossref]
- Vlachos A, Muir E (2010) How I treat Diamond-Blackfan anemia. Blood 116: 3715-3723. [Crossref]
- Dalle JH, Peffault de Latour R2 (2016) Allogeneic hematopoietic stem cell transplantation for inherited bone marrow failure syndromes. Int J Hematol 103: 373-379. [Crossref]
- Roy V, Pérez WS, Eapen M, Marsh JC, Pasquini M, et al. (2005) Bone marrow transplantation for diamond-blackfan anemia. Biol Blood Marrow Transplant 11: 600-608. [Crossref]
- Fagioli F, Quarello P, Zecca M, Lanino E, Corti P, et al. (2014) Haematopoietic stem cell transplantation for Diamond Blackfan anaemia: a report from the Italian Association of Paediatric Haematology and Oncology Registry. Br J Haematol 165: 673-681. [Crossref]
- Vlachos A, Federman N, Reyes-Haley C, Abramson J, Lipton JM (2001) Hematopoietic stem cell transplantation for Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Bone Marrow Transplant 27: 381-386. [Crossref]
- Farrar JE, Dahl N (2011) Untangling the phenotypic heterogeneity of Diamond Blackfan anemia. Semin Hematol 48: 124-135. [Crossref]
- Lipton JM, Ellis SR (2010) Diamond Blackfan anemia 2008-2009: broadening the scope of ribosome biogenesis disorders. Curr Opin Pediatr 22: 12-19. [Crossref]
- Moore E, Wisniewski A, Dobs Adrian (2011) Endocrine treatment of transsexual people: a review of treatment regimens, outcomes, and adverse effects. J Clin Endocrionol Metab 88: 3467-3473. [Crossref]
- Hamilton JB (1948) The role of testosterone secretion as indicated by the effects of castration in man and by studies of pathological conditions and the short lifespan associated with maleness. Recent Prog Horm Res 3: 257-289. [Crossref]
- Fonseca R, Rajkumar SV, White WL, Tefferi A, Hoagland HC (1998) Anemia after orchiectomy. Am J Hematol 59: 230-233. [Crossref]
- Grossmann M, Zajac JD (2012) Hematological changes during androgen deprivation therapy. Asian J Androl 14: 187-192. [Crossref]
- Calado RT, Young NS (2008) Telomere maintenance and human bone marrow failure. Blood 111: 4446-4455. [Crossref]
- Calado RT, Yewdell WT, Wilkerson KL, et.al. (2009) Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood 114: 2236-2243. [Crossref]
- Townsley DM, Dumitriu B, Liu D, Biancotto A, Weinstein B, et al. (2016) Danazol Treatment for Telomere Diseases. N Engl J Med 374: 1922-1931. [Crossref]
- Lansdorp PM (2016) Telomeres on Steroids--Turning Back the Mitotic Clock? N Engl J Med 374: 1978-1980. [Crossref]