Abstract
Several studies have indicated that exposure to pesticides is one of the main environmental factors that are associated with neurodegenerative diseases. Indeed, even the chronic exposure to low dosages of pesticides could be associated with some neurological diseases, including Alzheimer's disease (AD). In this review, we have discussed the mechanisms associated between exposure to pesticides and AD. We particularly concentrate on the impacts of pesticides on the oxidative stress, mitochondrial malfunction, formation of Aβ plaques, and hyper-phosphorylation of Tau proteins, which are important factors in the pathology of Alzheimer's disease.
Key words
pesticides, oxidative stress, aβ plaque formation, tau hyper-phosphorylation, Alzheimer’s disease.
Key message
Using pesticides in agriculture can lead to Alzheimer’s disease
Introduction
Alzheimer's disease is a brain disorder that gradually influences and decreases several mental abilities in humans. This chronic neurological disease usually has a slow course of progression. One of the most common early symptoms of AD is loss of short-term memory, and the progression symptoms can include speech problems, confusion, mood swings, self-care inability and behavioral issues [1]. However, the exact cause of AD is not known, it seems a multi-factorial process, which may include both genetic and environmental factors, impacting many cellular and molecular processes in the central nervous system(CNS) [2,3].
An exponential increase in the number of people suffering from AD since late 1990s indicates that environmental pollutions may have a significant impact on the incidence of this disease [4,5]. During the past few decades, synthetic insecticides and pesticides, which are toxic for the nervous system, have been extensively used worldwide [6,7]. Beyond affecting pests, these chemicals also affect non-target species and cause air, water and soil pollution [8]. Since most pesticides are fat-soluble and cannot be eliminated from the biological systems easily, they become more concentrated in higher levels of the food chain [9]. Thus, many of the commonly used pesticides remain in the body for a long time [10].
With respect to the organisms that pesticides target, they can be categorized as herbicides, insecticides, fungicides, and raticides [11]. In regards to their active sites, pesticides can be placed in the organochloride, organophosphate and carbamate groups. Chlorinated organic hydrocarbons, such as DDT[1], could divide into dichlorodiphenylethan and cyclodiene compounds. The toxicity level of these molecules varies depending on their degradation persistence and bioaccumulation potential [12].
A number of toxicology studies have shown that some pesticides, especially insecticides, such as Chlorpyrifos (Organophosphate insecticides), Cypermethrin (type II Pyrethroid insecticides), Maneb (Dithiocarbamate fungicides), and Warfarin (Rodenticides), can cause a considerable damage to the nervous system [13]. Exposure to high doses of insecticides (organophosphates and carbamate) can inhibit the Cholinesterase, which can lead to a continuous production of Acetylcholine, causing symptoms, such as weakness or paralysis [12]. Some organochloride insecticides and pyrethroids have neurotoxic effects on ion channels, which can disturb the balance of sodium/potassium in nerve fibers [14]. The basic function of the organophosphate pesticide is changing the neuronal signal transduction. Different pesticides have different action mechanisms. But, all of them eventually impact the transmission of nerve impulses.
High doses of pesticides can strongly affect the nervous system, but the question is how does exposure to low doses of these toxins affect the body?. Many reports have shown that in the areas where people were permanently exposed to pesticides, the prevalence of neurological diseases, such as AD, Parkinson’s Disease (PD), Multiple Sclerosis (MS) and polyneuropathy, was notably high [15-18]. Many studies have suggested that there might be a link between exposure to pesticides and the incidence of AD [19-22]. For example, a few epidemiology studies have shown that the prevalence of AD among some farmers who were exposed to organophosphate and organochloride was significantly higher compared to the rest of populations that had not experienced that exposure [21,23-25]. Singh and his colleagues have shown that use of organochloride pesticides (such as pp DDE[2], β- HCH[3], Dieldrin) and risk of AD are correlated [26], and that β-HCH levels in the serum of AD patients was several times higher compared to the healthy individuals. A study on 180 PD and AD patients who had the genetic risk factors for these diseases showed that individuals who had higher blood levels of the insecticide had a faster progression of AD and PD [27]. Another study reported that the serum levels of DDE[4] and DDT organochloride derivative compounds were remarkably higher in AD patients compared to the rest of population [28].
Exposure to pesticides at relatively low doses may affect the brain and lead to the loss of nerve cells in certain areas of the brain, which can have severe negative impacts on cognitive function, memory, advertence, mobility function in humans. These changes may eventually lead to AD [21,29].
Possible mechanisms underlying the impacts of pesticides on AD
Oxidative stress and mitochondrial dysfunction
Many human neurological diseases, such as AD, are related to the loss of mitochondrial function. The brain needs a lot of energy to maintain its normal physiological function. In addition, neurons are highly dependent on the energy that is produced in mitochondria [30,31]. Mitochondria are the primary consumers of oxygen and they contain multiple redox enzymes (oxidation/reduction) enabling them to transfer electron to oxygen molecules. Electron acceptor compounds are called oxidizing agents. The molecules that lose their electrons generate reactive oxygen species (ROS) [32]. ROS include reactive molecules and free radicals derived from molecules, which have oxygen atoms in their structures and have a short half-life, such as superoxide (O2-), hydroxyl (OH -),and hydrogen peroxide (H2O2) [33] (Figure 1)Furthermore, mitochondria play important roles in apoptosis. During the early stages of apoptosis, the potential difference in mitochondrial membrane is lost and some mitochondrial proteins, such as cytochrome c, are released into the cytosol [34].
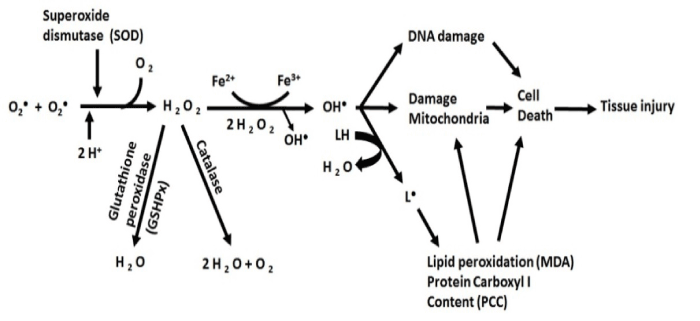
Figure 1. Effects of oxidative stress on cells: Lack of or excessive ROS production can interfere with the electron transfer change in mitochondria, which is a known risk factor in the pathology of Alzheimer’s disease.
Mitochondria are the main sites of ROS synthesis and have many antioxidant defense systems that can dispose the produced ROS. When mitochondria are damaged, the antioxidant system is compromised, resulting in an increased production of ROS and higher levels of oxidative stress, which leads to further mitochondrial damage. Imbalanced levels of cellular ROS leads to cellular damage and cell death [35,36]. ROS can be indirectly evaluated by measuring the levels of antioxidants and antioxidant enzymes. Some biochemical and cellular studies performed on postmortem brains of AD and AD transgenic mice have suggested that oxidative stress, mitochondrial damage and synaptic dysfunction are primary cellular changes in the development and progression of AD [37]. In fact, high levels of ROS have been reported to be correlated with the pathology of AD [38,39].
At the present time, most of the pesticides that are being used in many countries work based on toxic effects of oxidative stress , leading to an excessive production of reactive oxygen and nitrogen species (ROS, RNS), which can influence the level of antioxidants and inhibit the function of many cellular enzymes [40,41]. Some fungicides (e.g. Maneb) and insecticides (e.g. Rotenone) disturb neural functions through the suppression of mitochondrial function, which leads to an increased free radical production. Some herbicides also cause an uncoupled oxidative phosphorylation, which increases the levels of free radicals [42,43].
Exposure to pesticides, such as Zineb and Glyphosate, enhances the levels of oxidative stress in the brain and liver, which is correlated with the loss of cardiolipin and transmembrane potential content and simultaneously increases the peroxidation of fatty acids, especially in the substantia nigra [44,45]. To date, many studies have shown an increase in lipid peroxidation in AD [46-50] during which lipids are attacked by ROS, and chain reactions with free radicals lead to the production of lipid peroxidation products.
The impact of increased oxidative stress in aging and neurological degeneration is complicated. Oxidative stress is a result of imbalanced production and disposal (detoxification) of reactive oxygen species (ROS). ROS products are inevitable molecules that have a double-edged sword effect on biological systems. The balanced levels of ROS play crucial roles in maintaining many vital activities in cells, especially neurons. On the other hand, very high levels of ROS can lead to oxidation of important biomolecules, such as DNA, RNA, proteins and lipids, which can negatively affect cellular function and survival [51]. Several reports have shown that oxidative damages to DNA and RNA can cause rupture of double-stranded DNA, protein crosslinking, organic base alternations. Furthermore, a large number of DNA fractures have been found in the hippocampus and cortex of AD patients [52,53].
In addition to increased oxidative products in AD patient, serum levels of antioxidants, such as albumin, bilirubin, uric acid, lycopene, vitamin A, vitamin C and vitamin E are significantly reduced [54,55], and the activity of antioxidant enzymes, such as superoxide dismutase (SOD), catalase, glutathione peroxidase, and oxygenase in different brain regions of AD patients, including frontal and temporal cortex, are highly reduced [45,56,57].
Beta amyloid plaque formation
Many studies have indicated that the amyloid protein derived from amyloid precursor protein (APP) is the main factor in the pathogenesis of AD. APP is a transmembrane protein with unknown functions in different cell types, such as neurons [58]. This protein is attached to APP-like proteins, such as APL1 and APL2 [59]. When APP is proteolyzed by the enzymes β - or y - secretase, it can lead to the production of 38-43-amino acid peptides. Aβ molecules can form soluble, flexible and polymorphic oligomers. Aβ40 and Aβ42 isomers are the most common proteins [60]. But, accumulation of Aβ42 fibrils is more common in AD [61]. In the neurons of healthy individuals, the amounts of Aβ fragments are low and they can be easily lysed. But, overproduced Aβ fragments cannot be cleared in neurons, which can form spherical structures that are believed to play important roles in the pathogenesis of AD. Aβ plaques may have an impact on several age-related diseases by enhancing oxidative stress as well as increasing Aβ monomers or small oligomers, which can cause neuronal damage [62]. A specific isoform of apolipoproteins E (APOE4) is one of the important genetic risk factors in AD. The pathological interactions between this protein and APP has been shown to be correlated with AD [63]. Moreover, some mutation in the triggering receptor expressed on myeloid cells 2 (TREM2) gene are a risk factor for developing AD. The normal interaction between the TREM2 protein and microglia in the brain is necessary for the activation of these cells to clean up plaques. When this gene is mutated, it can interfere with its normal effect on the mentioned microglia’s cleaning function, increasing the chance of developing AD [64]. Aβ is produced in the perikarya of neurons and transmitted to the axons terminal. Recent studies have shown that Aβ can accumulate in the mitochondrial membrane and induce the production of free radicals. This phenomenon can reduce the activity of mitochondrial complex IV. As a result, the production of ATP is reduced, resulting in mitochondrial dysfunction and neuronal death [65]. Many studies have suggested that neuronal damage, synapsis failure and neurofibrillary network (NFT) are pathological symptoms of AD [66,67] (Figure 2).
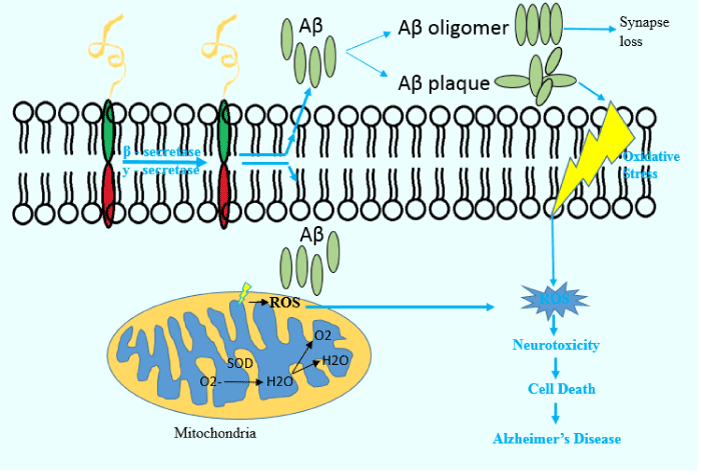
Figure 2. Aβ plaques and oxidative stress. β - and y – secretase cleave APPs and produce Aβ units. Outside cells, these proteins can lead to synapse loss and oxidative stress. Inside cells, Aβ units can increase the generation of ROS, causing neurotoxicity and cell death.
Long term exposure to some organophosphate pesticides, such as Chlorpyrifos, increase beta-amyloid levels in the brain [68]. Organic chlorinated pesticides are fat-soluble toxins that remain stable in the environment and food chains. In humans, hemostatic factors are stored between organochloride toxins in adipose tissues and their concentration in the blood circulation is not clear [69]. Although these toxins are consistently stored in fat tissues, they can be released in the blood after a rapid weight loss [70]. According to Jason Richardson, long-term exposure to toxins, such as DDT and DDE, can increase the levels of APP, which is correlated to the formation of Aβ plaques. Moreover, in AD patients who often lose weight, the release of such organochlorinated factors in the blood accelerates the progression of AD [28].
Tau Phosphorylation
Biochemical and cellular studies on rats continuously exposed to pesticides, such as Carbofuran and deltamethrin, have shown an excessive tau phosphorylation occurring by the activation of glycogen synthase kinase-3β and inhibition of protein phosphatase-2A [71]. Tau phosphorylation leads to the formation of the two stranded and intertwined neurofibrils in the nerve cells [72], which can lead to the disintegration of microtubules, loss of cellular structure and malfunction in the neural functions [73]. At first, it may disrupt the biochemical communication between neurons followed by cell death [74] (Chun W 2007). Aβ aggregation and phosphorylated tau can block cell organelle transfer, including mitochondria, to synaptic terminals where high ATP is required. For instance, in a study using neuronal and non-neuronal cells with multiple Tau mutants, this protein was able to block the backward movements of vesicles and reduce cell organelle transportation by blocking microtubules. Tau was also involved in reducing mitochondrial ATP in dendrites and synapses [75]. In addition, another study on the effect of Tau on vesicle and organell transportation in primary cortical neurons, retinal ganglion cells and neuroblastoma showed that Tau inhibited the kinesin-dependent transmission of peroxisomes, neurofilament (NF) and derivative vesicles in these cells. The absence of peroxisomes makes the cells vulnerable to oxidative stress and cause neuronal damage. This study also indicated that Tau blocks APP transfer to axons and dendrites and interferes with the axonal transport [76].
Conclusion
The use of pesticides for pest control has become common around the world. Pesticides are found in the air we breathe, the water we drink and the food we eat. Thus, pesticides are associated with a wide spectrum of health problems, including neurological diseases, especially AD. Exposure to low doses of pesticides for a long time can enhance the formation of AB plaques, resulting in the increased level of oxidative stress. ROS produced in cytoplasm reduce the expression levels of antioxidants, leading to cell death. ROS produced in the mitochondria and intracellular tau filaments are able to create neuronal toxicity. Eventually, neural death occurs. All these pathologic phenomena can lead to or accelerate AD. Further research is clearly required to shed light on the mechanisms in which pesticides affect the cellular risk factors of AD. The present review underlines the necessity of designing pesticides with zero side effects on the human body, especially the nervous system, as well as developing other methods of dealing with pests, such as using biological pest control and insect breeding interference- based genetic engineering practices.
Acknowledgments
We thank Amanda Marie Di Battista from Dr. G. William Rebeck’s lab at Georgetown University for her constructive comments on our manuscript.
Conflict of interest
The authors reveal no conflict of interest.
Author contribution
EG, MT, PP, SF wrote the manuscript. NGA and MT edited the manuscript.
[1] Dichlorodiphenyltrichloroethane
[2]p,p′-Dichlordiphenyldichlorethen
[3]β-Hexachlorocyclohexane
[4]Dichlorodiphenyldichloroethylene
References
References
- Burns A IS (2009) Alzheimer's disease. BMJ (Clinical research ed). 338: 158. [Crossref]
- D G (1987). Le paradigme de la maladied'Alzheimer.
- Thomas P, Fenech M (2007) A review of genome mutation and Alzheimer's disease. Mutagenesis 22: 15-33. [Crossref]
- Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, et al. (2005) Global prevalence of dementia: a Delphi consensus study. The Lancet 366: 2112-2117. [Crossref]
- Ramaroson H, Helmer C, Barberger-Gateau P, Letenneur L, Dartigues JF, et al. (2003) Prevalence of dementia and Alzheimer's disease in people aged 75 and over: Updated the PAQUID data. Rev Neurol (Paris) 4: 405-411. [Crossref]
- Casida JE, Quistad QG (2004) Why insecticides are more toxic to insects than people: the unique toxicology of insects. J Pestic Sci 29: 81-86.
- Clive D.S. Tomlin (2009) The Pesticide Manual: A World Compendium, 15 edn, Hampshire, UK.
- GT M (2004). Sustaining the Earth, vol Chapter 9, Thompson Learning, Incedn. Pacific Grove, California
- Castro P ME (2010) Marine Biology. McGraw-Hill Companies Inc, New York
- D Pimentel (1995) Amounts of pesticides reaching the target pests. J Agric Environ Ethics 8: 17-22.
- Gilden RC, Huffling K, Sattler B (2010) Pesticides and health risks. J Obstet Gynecol Neonatal Nurs 39: 103-110. [Crossref]
- Michael A. Kamrin (1997) Pesticide Profiles: toxicity, environmental impact, and fate. CRC Press.
- Bjørling-Poulsen M, Andersen HR, Grandjean P (2008) Potential developmental neurotoxicity of pesticides used in Europe. Environ Health 7: 50. [Crossref]
- Keifer MC, Firestone J (2007) Neurotoxicity of pesticides. J Agromedicine 12: 17-25. [Crossref]
- Manthripragada AD, Costello S, Myles GC, Bronstein JM, Ritz B (2010) Paraoxonase 1 (PON1).agricultural organophosphate exposure, and Parkinson disease. Epidemiology 21: 87-94. [Crossref]
- Kamel F, Hoppin JA (2004) Association of pesticide exposure with neurologic dysfunction and disease. Environ Health Perspect 112: 950-958. [Crossref]
- Sordet-Guépet, Hélène; Manckoundia, Patrick (2014) Alzheimer disease and environmental exposure to neurotoxic factors. A controversy. Br J Med Med Res 34: 5298-5311.
2021 Copyright OAT. All rights reserv
- Parrón T, Requena M, Hernández AF, Alarcón R (2011) Association between environmental exposure to pesticides and neurodegenerative diseases. Toxicol Appl Pharmacol 256: 379-385. [Crossref]
- Baldi I,Lebailly P, Mohammed-Brahim B, Letenneur L, Dartigues JF, et al. (2003) Neurodegenerative diseases and exposure to pesticides in the elderly. Am J Epidemiol 157: 409-414. [Crossref]
- Baltazar MT, Dinis-Oliveira RJ, de Lourdes Bastos M, Tsatsakis AM, Duarte JA, et al. (2014) Pesticides exposure as etiological factors of Parkinson's disease and other neurodegenerative diseases. Toxicol Lett 230: 85-103. [Crossref]
- Hayden KM, Norton MC, Darcey D, Ostbye T, Zandi PP, et al. (2010) Occupational exposure to pesticides increases the risk of incident AD. Neurology 74: 1524-1530. [Crossref]
- Tyas SL, Manfreda J, Strain LA, Montgomery PR (2001) Risk factors for Alzheimer’s disease: a population-based, longitudinal study in Manitoba, Canada. Int J Epidemiol 30: 590-597. [Crossref]
- Casida JE, Durkin KA (2013) Neuroactive insecticides: targets, selectivity, resistance, and secondary effects. Annu Rev Entomol 58: 99-117. [Crossref]
- Chen L, Yoo SE, Na R, Liu Y, Ran Q (2012) Cognitive impairment and increased Aβ levels induced by paraquat exposure are attenuated by enhanced removal of mitochondrial H(2)O(2). Neurobiol Aging 432: 15-26. [Crossref]
- Zaganas I, Kapetanaki S, Mastorodemos V, Kanavouras K, Colosio C, et al. (2013) Linking pesticide exposure and dementia: what is the evidence? Toxicology 307: 3-11. [Crossref]
- Singh N, Chhillar N, Banerjee B, Bala K, Basu M, et al. (2013) Organochlorine pesticide levels and risk of Alzheimer’s disease in north Indian population. Hum Exp Toxicol 32: 24-30. [Crossref]
- Chhillar N, Singh NK, Banerjee B, Bala K, Basu M (2012). Beta hexachlorocyclohexane (β-HCH).and Risk of Alzheimer’s Disease and Parkinson’s Disease. International Conference on Biological and Life Sciences: 40.
- Richardson JR, Roy A, Shalat SL, von Stein RT, Hossain MM, et al. (2014) Elevated Serum Pesticide Levels and Risk for Alzheimer Disease. JAMA Neurol 71: 284-290. [Crossref]
- Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, et al. (2011) Rotenone, Paraquat and Parkinson's Disease. Environ Health Perspect 119: 866-872. [Crossref]
- Chong ZZ, Li F, Maiese K (2005) Oxidative stress in the brain: novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol 75: 207-246. [Crossref]
- Moreira PI, Duarte AI, Santos MS, Rego AC, Oliveira CR (2009) An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer’s disease. J Alzheimers Dis 16: 741-761. [Crossref]
- Cheeseman KH, Slater TF (1993). An introduction to free radical biochemistry. Br Med Bull 49: 481-493. [Crossref]
- Haber F, Weiss J (1932) Über die Katalyse des Hydroperoxydes [On the catalysis of hydroperoxides]. Naturwissenschaften 20: 948-950.
- Pope S, Land JM, Heales SJ (2008) Oxidative stress and mitochondrial dysfunction in neurodegeneration; cardiolipin a critical target? Biochim Biophys Acta 1777: 794-799. [Crossref]
- Di Carlo M, Giacomazza D, Picone P, Nuzzo D, San Biagio PL (2012) Are oxidative stress and mitochondrial dysfunction the key players in the neurodegenerative diseases? Free Radic Res 46: 1327-1338. [Crossref]
- Picone P, Bondi ML, Montana G, Bruno A, Pitarresi G, et al. (2009) Ferulic acid inhibits oxidative stress and cell death induced by Ab oligomers: improved delivery by solid lipid nanoparticles. Free Radic Res 43: 1133-1145. [Crossref]
- Lee J, Giordano S, Zhang J (2012) Autophagy, mitochondria and oxidative stress: cross-talk and redox signaling. J Biochem 441: 523-540. [Crossref]
- Dumont M, Beal MF (2011) Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic Biol Med 5: 1014-1026. [Crossref]
- Müller M, Cheung KH, Foskett JK (2011) Enhanced ROS generation mediated by Alzheimer’s disease presenilin regulation of InsP3R Ca2+ Antioxid Redox Signal 7: 1225-1235. [Crossref]
- Beuret CJ, Zirulnik F, Gimenez MS (2005) Effect of the herbicide glyphosate on liver lipoperoxidation in pregnant rats and their fetuses. Reprod Toxicol 19: 501-504. [Crossref]
- Sharma Y, Bashir S, Irshad M, Gupta SD, Dogra TD (2005) Effects of acute dimethoate administration on antioxidant status of liver and brain of experimental rats. Toxicology 206: 49-57. [Crossref]
- Witold Seńczuk (2005) Toksykologiawspolczesna. PZWL, Warszawa.
- Wang X, Wang W, Li L, Perry G, Lee HG, et al. (2014) Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta 1842: 1240-1247. [Crossref]
- Astiz M, de Alaniz MJ, Marra CA (2009) Effect of pesticides on cell survival in liver and brain rat tissues. Ecotoxicol Environ Saf 72: 2025-2032. [Crossref]
- Venkateshappa C, Harish G, Mahadevan A, Srinivas Bharath MM, Shankar SK (2012) Elevated oxidative stress and decreased antioxidant function in the human hippocampus and frontal cortex with increasing age: implications for neurodegeneration in Alzheimer's disease. Neurochem Res 37: 1601-1614. [Crossref]
- Ferreira PM, Militão GC, Freitas RM (2009) Lipoic acid effects on lipid peroxidation level, superoxide dismutase activity and monoamines concentration in rat hippocampus. Neurosci Lett 464: 131-134. [Crossref]
- Militão GC, Ferreira PM, de Freitas RM (2010) Effects of lipoic acid on oxidative stress in rat striatum after pilocarpine-induced seizures. Neurochem Int 56: 16-20. [Crossref]
- McGrath LT, McGleenon BM, Brennan S, McColl D, McILroy S, et al. (2001) Increased oxidative stress in Alzheimer's disease as assessed with 4-hydroxynonenal but not malondialdehyde. QJM 94: 485-490. [Crossref]
- DiCiero Miranda M, de Bruin VM, Vale MR, Viana GS (2000) Lipid peroxidation and nitrite plus nitrate levels in brain tissue from patients with Alzheimer's disease. Gerontology 46: 179-184. [Crossref]
- Montine KS, Reich E, Neely MD, Sidell KR, Olson SJ, et al. (1998) Distribution of reducible 4-hydroxynonenal adduct immunoreactivity in Alzheimer disease is associated with APOE genotype. J Neuropathol Exp Neurology 57: 415-425. [Crossref]
- Ferrer MD, Sureda A, Mestre A, Tur JA, Pons A (2010) The double edge of reactive oxygen species as damaging and signaling molecules in HL60 cell culture. Cell Physiol Biochem 25: 241-252. [Crossref]
- Anderson AJ, Su JH, Cotman CW (1996) DNA damage and apoptosis in Alzheimer's disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J Neurosci 16: 1710-1719. [Crossref]
- Mullaart E, Boerrigter ME, Ravid R, Swaab DF, Vijg J (1990) Increased levels of DNA breaks in cerebral cortex of Alzheimer's disease patients. Neurobiol Aging 11: 169-173. [Crossref]
- Foy CJ, Passmore AP, Vahidassr MD, Young IS, Lawson JT (1999) Plasma chain breaking antioxidants in Alzheimer's disease, vascular dementia and Parkinson's disease. QJM 92: 39-45. [Crossref]
- Kim TS, Pae CU, Yoon SJ, Jang WY, Lee NJ, et al. (2006) Decreased plasma antioxidants in patients with Alzheimer's disease. Int J Geriatr Psychiatry 21: 344-348. [Crossref]
- Marcus DL, Thomas C, Rodriguez C, Simberkoff K, Tsai JS, et al. (1998) Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer's disease. Exp Neurol 150: 40-44. [Crossref]
- Omar RA, Chyan YJ, Andorn AC, Poeggeler B, Robakis NK, et al. (1999) Increased expression but reduced activity of antioxidant enzymes in Alzheimer's disease. J Alzheimers Dis 1: 139-145. [Crossref]
- Lee SH, Kim Y, Kim HY, Kim YH, Kim MS, et al. (2014) Aminostyrylbenzofuran directly reduces oligomeric Amyloid-ß and Reverses Cognitive Deficits in Alzheimer transgenic mice. PLoS One 9: 57-63. [Crossref]
- Blennow K, de Leon MJ, Zetterberg H (2006). Alzheimer›s disease. Lancet 368: 387-403. [Crossref]
- Hartmann T, Bieger SC, Brühl B, Tienari PJ, Ida N, et al. (1997) Distinct sites of intracellular production for Alzheimer's disease A beta40/42 amyloid peptides. Nat Med 3: 1016-1020. [Crossref]
- Yin YI,BassitB, Zhu L, Yang X, Wang C, et al. (2007) γ-Secretase Substrate Concentration Modulates the Aβ42/Aβ40 Ratio: Implications for Alzheimer's disease. J Biol Chem 282: 23639-23644.
- Ashabi G, Ahmadiani A, Abdi A, Abraki SB, Khodagholi F (2013) Time course study of Aß formation and neurite outgrowth disruption in differentiated human neuroblastoma cells exposed to H2O2: protective role of autophagy. Toxicol In Vitro 6: 1780-1788. [Crossref]
- Polvikoski T, Sulkava R, Haltia M, Kainulainen K, Vuorio A, et al. (1995) Apolipoprotein E, Dementia, and Cortical Deposition of Beta-amyloid Protein. N Engl J Med 333: 1242-1247. [Crossref]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, et al. (2012) Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368: 107-117. [Crossref]
- Rosales-Corral S, Acuna-Castroviejo D, Tan DX, López-Armas G, Cruz-Ramos J, et al. (2012). Accumulation of exogenous amyloid-beta peptide in hippocampal mitochondria causes their dysfunction: a protective role for melatonin. Oxidative Medicine and Cellular Longevity: 15.
- Geodert M, Spillantini MG (2006) A Century of Alzimer`s disease. Science 314: 777-781. [Crossref]
- Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N Engl J Med 362: 329-344. [Crossref]
- Salazar JG, Ribes D, Cabré M, Domingo JL, Sanchez-Santed F, et al. (2011) Amyloid β peptide levels increase in brain of AβPP Swedish mice after exposure to chlorpyrifos. Curr Alzheimer Res 8: 732-740. [Crossref]
- Sandanger TM, Sinotte M, Dumas P, Marchand M, Sandau C, et al. (2007) Plasma concentrations of selected organobromine compounds and polychlorinated biphenyls in postmenopausal women of Quebec, Canada. Environ Health Perspect 115: 1429-1434. [Crossref]
- De Roos AJ, Ulrich CM, Sjodin A, McTiernan A (2012) Adiposity body composition, and weight change in relation to organochlorine pollutant plasma concentrations. J Expo Sci Environ Epidemiol 22: 617-624. [Crossref]
- Chen NN, Luo DJ, Yao XQ, Yu C, Wang Y, et al. (2012) Pesticides induce spatial memory deficits with synaptic impairments and an imbalanced tau phosphorylation in rats. J Alzheimers Dis 30: 585-594. [Crossref]
- Goedert M, Spillantini M, Crowther RA (1991) Tau proteins and neurofibrillary degeneration. Brain Pathol 1: 279-286. [Crossref]
- Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, et al. (2005) Tau pathology in alzheimer disease and other tauopathies. Biochim Biophys Acta 1739: 198-210. [Crossref]
- Chun W, Johnson GV (2007) The Role of The role of tau phosphorylation and cleavage in neuronal cell death. Front Biosci 12: 733-756. [Crossref]
- Eckert A, Hauptmann S, Scherping I, Rhein V, Müller-Spahn F, et al. (2008) Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener Dis 5: 157-159. [Crossref]
- Thies E, Mandelkow EM (2007) Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci 11: 2896-2907. [Crossref]