Human uterine leiomyosarcoma (U-LMS) is neoplastic malignancy that typically arises in tissues of mesenchymal origin. The identification of novel molecular mechanism leading to human U-LMS formation and the establishment of new therapies has been hampered by several critical points. We earlier reported that mice with a homozygous deficiency for proteasome beta subunit 9 (Psmb9)/β1i, an interferon (IFN)-γ inducible factor, spontaneously develop U-LMS. The use of research findings of the experiment with mouse model has been successful in increasing our knowledge and understanding of how alterations, in relevant oncogenic, tumour suppressive, and signaling pathways directly impact sarcomagenesis. The IFN-γ pathway is important for control of tumour growth and invasion and has been implicated in several malignant tumours. In this study, experiments with human tissues revealed a defective expression of PSMB9/β1i in human U-LMS that was traced to the IFN-γ pathway and the specific effect of somatic mutations of JANUS KINASE (JAK) 1 molecule or promoter region on the locus cording PSMB9/β1i gene. Understanding the molecular mechanisms of human U-LMS may lead to identification of new diagnostic candidates or therapeutic targets against human U-LMS.
PSMB9/β1i, IFN-γ, somatic mutation, uterine leiomyosarcoma
Human uterine leiomyosarcoma (U-LMS) is neoplastic malignancy that typically arises in tissues of mesenchymal origin. The identification of novel molecular mechanism leading to human U-LMS formation and the establishment of new therapies has been hampered by several critical points. We earlier reported that mice with a homozygous deficiency for proteasome beta subunit (Psmb)9/β1i, an interferon (IFN)-γ inducible factor, spontaneously develop U-LMS. The use of research findings of the experiment with mouse model has been successful in increasing our knowledge and understanding of how alterations, in relevant oncogenic, tumour suppressive, and signaling pathways directly impact sarcomagenesis. The IFN-γ pathway is important for control of tumour growth and invasion and has been implicated in several malignant tumours. In this study, experiments with human tissues revealed a defective expression of PSMB9/β1i in human U-LMS that was traced to the IFN-Γ pathway and the specific effect of somatic mutations of JANUS KINASE (JAK) 1 molecule or promoter region on the locus cording PSMB9/β1i gene. Understanding the molecular mechanisms of human U-LMS may lead to identification of new diagnostic candidates or therapeutic targets against human U-LMS.
Uterine mesenchymal tumours have been traditionally divided into benign tumour leiomyomas (LMA) and malignant tumour leiomyosarcomas (LMS) based on cytological atypia, mitotic activity and other criteria. Uterine LMS (U-LMS), which are some of the most common neoplasms of the female genital tract, are relatively rare uterine mesenchymal tumour, having an estimated annual incidence of 0.64 per 100,000 women [1]. They account for approximately one-third of uterine sarcomas, of only 53% for tumours confined to the uterus [2,3]. Generally, patients with U-LMS typically present with vaginal bleeding, pain, and a pelvic mass. Gynecological tumour, for instance breast cancer and endometrial carcinomas, are strongly promoted by female hormones, but the rate of hormone receptor expression is reported to be significantly less in human U-LMS compared with normal myometrime. These low expressions of receptor were found to not correlate with the promotion of initial disease development or with the overall survival of patients with U-LMS.
As U-LMS is resistant to chemotherapy and radiotherapy, and thus surgical intervention is virtually the only means of treatment for this disease, however, molecular targeting therapies against tumours have recently shown remarkable achievements [4-8]. It is noteworthy that, when adjusting for stage and mitotic count, LMS has a significantly worse prognosis than carcinosarcoma; developing an efficient adjuvant therapy is expected to improve the prognosis of the disease [9]. A trend towards prolonged disease-free survival is seen in patients with matrix metalloproteinase (MMP)-2-negative tumours [10]. Although typical presentations with hypercalcemia or eosinophilia have been reported, this clinical abnormality is not an initial risk factor for U-LMS. To the best of our knowledge, little is known regarding the biology of U-LMS; therefore, the risk factors that promote the initial development of U-LMS and regulate their growth in vivo remain poorly understood.
The mice with a targeted disruption of proteasome beta subunit 9 (PSMB9)/β1i, which is interferon (IFN)-γ-inducible proteasome subunit, exhibited a defect in tissue- and substrate- dependent proteasome function, and female PSMB9/β1i-deficient mice shown to develop U-LMS, with a disease prevalence of 37% by 14 months of age [11,12]. Defective expression of PSMB9/β1i is likely to be one of the risk factors for the development of human U-LMS, as it is in PSMB9/β1i-deficient mice [12]. Recent report shows that stable PSMB9/β1i expression contributes to cell proliferation, which directly correlates to the progressive deterioration with increasing stage and grade of the tumour. As the importance and involvement of the IFN-γ pathway in the activation of shared-promoter of the transporter associated with antigen processing (TAP) 1 and PSMB9/β1i have been established, it is demonstrated that the defective expression of PSMB9/β1i was attributable to G871E somatic mutation in the ATP-binding region of JAK1 in SKN cell line, which is established from patient with U-LMS. It is furthermore likely that the PSMB9/β1i expressions are down-regulated in human U-LMS tissues such like human U-LMS cell line. We demonstrate that there are serious mutational defects in the factors on the IFN-γ pathway, which is the key cell-signaling pathway for PSMB9/β1i expression and promoter region of PSMB9/β1i gene, in human U-LMS. The somatic mutational defects in the IFN-γ pathway may induce the initial development of U-LMS. Recent advances in our understanding of the biological characters of U-LMS have concentrated on the impaired IFN-γ pathway. It is clear that somatic mutations in key regulatory genes alter the behavior of cells and can potentially lead to the unregulated growth seen in malignant tumour. Therefore, continued improvement of our knowledge of the molecular biology of U-LMS may ultimately lead to novel therapies and improved outcome.
The effects of IFN-γ on expression of PSMB9/β1i was examined using five cell lines [13]. Expressions of PSMB9/β1i were not markedly induced by IFN-γ treatment in human U-LMS cell lines, although cervical epithelial adenocarcinoma cell lines and normal human uterus smooth muscle cells underwent strong induction of PSMB9/β1i following IFN-γ treatment [13]. Furthermore, the immunohistochemistry (IHC) experiments revealed a serious loss in the ability to induce expression of PSMB9/β1i in human U-LMS tissues in comparison with normal myometrium tissues located in same tissue sections and other 4 mesenchymal tumour types. Of 58 U-LMS, 50 cases were negative for PSMB9/β1i, 4 cases were focally positive, 2 cases were weakly positive, and 2 cases were positive. IHC analyses showed positivity for Ki-67/MIB1 and differential expression of ESTROGEN RECEPTOR (ER), PROGESTERONE RECEPTOR (PR), TUMOUR PROTEIN 53 (TP53), and CALPONIN h1. In addition, the expression level of PSMB9/β1i was also examined in the skeletal muscle metastasis from U-LMS, the histological diagnosis was consistent with metastatic LMS for skeletal muscle lesions. Pathological study of surgical samples showed presence of a mass measuring 3 cm at largest diameter in lumbar quadrate muscle without a fibrous capsule. All lymph nodes were negative. In western blotting and RT-PCR experiments, PSMB9/β1i was expressed in normal myometrium, LMA, and IFN-Γ-treated HeLa cells, but not in human U-LMS. The both research experiments strongly supported the research findings obtained from IHC experiments.
Most frequently, LMS have appeared in the uterus, retroperitoneum or extremities, and although histologically indistinguishable, they have different clinical courses and chemotherapeutic responses. The molecular basis for these differences remains unclear. Therefore, the examination of human U-LMS tissues (23 U-LMS tissue sections and normal tissue sections located in the same tissue) was performed to detect somatic mutations in the IFN-γ pathway, JAK1, JAK2, STAT1 and promoter region of PSMB9/β1i gene (Figure 1). As the catalytic domains of these factors are most likely to harbour mutations that activate the gene product, we focused on stretches (exons) containing the kinase domains, transcriptional activation domains and enhancer/promoter region. Over all, nearly 43.5% (10/23) of U-LMS tissues had serious mutations in the ATP binding region or kinase-specific active site of JAK1; furthermore, 43.5% (10/23) of U-LMS tissues had serious mutations in essential sites of the promoter region of PSMB9/β1i gene, which is required for transcriptional activation of PSMB9/β1i gene (Table 1). No somatic mutation in essential sites, Tyr701 and Ser727, which are required for STAT1 transcriptional activation, was elucidated in uterine LMS. Nearly 21.7% (5/23) of U-LMS tissues unexpectedly had mutations in the STAT1 intermolecular region, which is not yet reported to be important for biological function as transcriptional activation. No somatic mutation in the ATP-binding region and kinase-active site of JAK2 was detected in U-LMS (Table 1). MOTIF Search profiling [14] and NCBI's Conserved Domain Database and Search Service, v2.17 analysis also revealed that somatic mutations, which were identified in the catalytic domains of these genes, resulted in impaired activations of tyrosine kinases or transcriptional factor [15].
Table 1. Somatic mutations in IFN-γ signaling pathway in human uterine leiomyosarcoma. The data of somatic mutations in table 1 was shown separately with respect to each gene, JAK1, JAK2, STAT1 and activation region of the promoter of PSMB9/β1i gene.
Mutations in the IFN-y pathway in human uterine Ieiomyosarcoma |
Gene Name |
Locus |
GenBank Accession |
MIM ID |
Tumor |
Nucleotide |
Amino Acid |
Domain |
Evolutionary
conservation |
JAK1 |
HUMPTKJAK1 |
M64174.1 |
*147795 |
ULMS |
G2612A
G2618A
G2626A
G2642T
G2643A
A2957C
A2960C
A2985T |
G781E
G873D
G876R
C881F
C881 Stop Q986P
Y987S
R995S |
ATP binging
ATP binding
ATP binging
ATP binging
ATP binding
active site
active site
active site |
p,c,m,r,g,d |
JAK2 |
AF005216 |
AF005216.1 |
+147796 |
ULMS |
ND2 |
ND |
ND |
p,c,b,m,r,g,d |
STAT1 |
NM_007315 |
NM_007315 |
+600555 |
ULMS |
A2104C
T2128G
T2078G
A2148C |
1702E
S710A
L693R
R7168 |
NA3
NA
NA
NA |
c,b,m,r,g,d |
PSMB91 |
X62741 |
X62741.1 |
*177045 |
ULMS |
A209T
A210G
C213A
C214T
G215A
A216G
A217G
G219 A
G239 A |
|
RF-E site
RF-E site
RF-E site
RF-E site
RF-E site
RF-E site
RF-E site
RF-E site
HSF site |
|
2021 Copyright OAT. All rights reserv
1LMP2 promoter region, NCBI Reference Sequence NT_007592.15 Homo sapiens Chromosome 6 2not detected 3non-kinase activation region 4Evolutionary conservation refers to the species in which an identical residue was observed in the homolog (p, pan troglodytes; c, canis lupus familiaris; b, bos taurus; m, mus musculus; r, rattus norvegicus; g, gallus gallus; d, danio rerio)
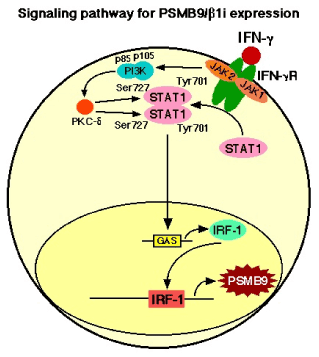
Figure 1. IFN-γ treatment markedly increased the expression of PSMB9/β1i, a subunit of the immunoproteasome, which alters the proteolytic specificity of proteasomes. After binding of IFN-γ to the type II IFN receptor, which is constructed by two components, IFN-γ receptor subunit 1 (IFNGR1) and IFN-γ receptor subunit 2 (IFNGR2), Janus-activated kinase 1 (JAK1) and JAK2 are activated and phosphorylate the signal transducer and activator of transcription 1(STAT1) on the tyrosine residue at position 701 (Tyr701) and the serine residue at position 727 (Ser727) [32,33]. Tyrosine phosphorylated STAT1 forms homodimers that translocate to the nucleus and bind GAS (IFN-γ-activated site) elements in the promoters of IFN-γ-regulated genes [32,33]. IFN-γ activated JAKs also regulate, through as yet unknown intermediates, activation of the catalytic subunit (p110) of phosphatidylinositol 3-kinase (PI3K). The activation of PI3K ultimately results in downstream activation of protein kinase C-δ (PKC-δ), which in turn regulates the phosphorylation of STAT1 on the Ser727. The phosphorylation of Ser727 is not essential for the translocation of STAT1 to the nucleus or for the binding of STAT1 to enhancer/promoter region of targeted DNA, but it is required for full transcriptional activation [34,35].
In a recent report, a comparative genomic hybridization (CGH)-based analysis of LMS using a high-resolution genome-wide array gave gene-level information about the amplified and deleted regions that may play a role in the development and progression of human U-LMS. Other reports showed that among the most intriguing changes in genes were losses of JAK1 (1p31-p32) and PSMB9/β1i (6p21.3) [16,17]. It has also been demonstrated that a correlation exists between the development of malignant tumours and ethnic background, so we conducted CGH experiments with tissue samples obtained from Japanese patients in order to obtain gene-level information. Our results showed that LMS having a clear functional loss at JAK1 (1p31-p32) and PSMB9/β1i (6p21.3) also harbored one nonsense mutation and one deletion, suggesting a possible homozygous loss of function. The discovery of these mutational defects in a key cell-signaling pathway may be important in understanding the pathogenesis of human U-LMS.
Uterine LMS are relatively rare mesenchymal tumours, having an estimated annual incidence of 0.64 per 100 000 women. They account for approximately one-third of uterine sarcomas and 1.3% of all uterine malignancies. They are the disease with extremely poor prognosis, considering aggressive malignancies with a 5-year survival rate of only 50% for tumours confined to the uterus. At present, surgical intervention is virtually the only means of treatment for U-LMS [4-8]. Although adjuvant pelvic irradiation appears to decrease the rate of local recurrence, adjuvant therapy does not appear to significantly improve survival. Furthermore, gynecological cancer, for instance breast cancer and endometrial carcinomas, are strongly promoted by female hormones, but the rate of estrogen receptor and progesterone receptor expression is reported to be significantly less in human U-LMS compared with normal myometrium. These low receptor expressions were found to not correlate with the promotion of initial disease development or with the overall survival of patients with U-LMS; however, molecular targeting therapies against tumours have recently shown remarkable achievements [18]. To improve the prognosis of human U-LMS, research experiments were performed to identify the key role of pro- or anti-oncogenic factors that have an important function in their pathogenesis and that could serve as molecular targets for tumour treatment. For this purpose, several research facilities conducted a microarray procedure between human U-LMS and normal myometrium and showed that several known pro-oncogenic factors, such as brain-specific polypeptide PEP-19 and a transmembrane tyrosine kinase receptor, c-KIT, may be associated with the pathogenesis of human U-LMS [19-21]. However, in terms of the tumourigenesis of human U-LMS, merely comparing the expression of potential pro-oncogenic factors between normal and malignant tissues is not sufficient because the results obtained may be the consequence of malignant transformation and, therefore, not necessarily the cause. In addition, dysregulation of apoptotic mechanisms has also been implicated in many human malignancies. Although the significant differential expression of apoptotic and cell cycle regulatory factors in human U-LMS, such as B-cell Lymphoma-2 (BCL-2), BCL-2-Associated X protein (BAX), P16 Inhibits CDK4 (P16/INK4a), P21 Cyclin-Dependent Kinase Inhibitor 1 (P21/CIP1), P27 Kinase Inhibitor Protein 1 (P27/KIP1), Cellular v-KIT Hardy-Zuckerman 4 Feline Sarcoma Viral Oncogene Homolog (c-KIT), Mitogen-Inducible Gene-2 (MIG-2), MDM2, TUMOUR PROTEIN 53 (TP53), have all been reported and compared to normal myometrium, there exists no scientific evidence to show that abnormal expression of these factors directly correlates to the initiation and promotion of human U-LMS. PSMB9/β1i-dificient mice were reported to be prone to the development of U-LMS, but not in their parental mice, C57BL/6 mice [12]. The percentage of mice with overt tumours increased with age after six months, with a cumulative prevalence of disease in female mice of 37% by 14 months of age and no apparent plateau at this late observation time. Histopathological examinations of PSMB9/β1i-deficient uterine neoplasms revealed common characteristic abnormalities of U-LMS. In addition, recent research reports show the loss in the IFN-Γ-inducible ability of PSMB9/β1i expressions in SKN cell line and other primary U-LMS cells established from patients. The histopathological experiments demonstrated a serious loss in the ability to induce the expression of PSMB9/β1i in human U-LMS tissues in comparison with normal myometrim tissues located in same tissue sections.
IFN-Γ treatment markedly increased the expression of PSMB9/β1i, a subunit of the proteasome, which alters the proteolytic specificity of proteasomes. Sequence analysis demonstrated that the loss of IFN-g responsiveness in the human U-LMS cell line was attributable to the inadequate kinase activity of JAK1 due to a G781E somatic mutation in the ATP-binding region [13]. The defect was localized to JAK1 activation, which acts upstream in the IFN-γ signal pathway since IFN-γ treatment could not strongly induce JAK1 kinase activity in human U-LMS cell lines. Genetic alterations in tyrosine kinases have previously been firmly implicated in tumourigenesis, but only a few serine/threonine kinases are known to be mutated in human cancers [22-25]. For instance, mice carring homozygous deletion of Phosphatase and Tensin Homolog Deleted from Chromosome 10 (Pten) alleles developed widespread SMC hyperplasia and abdominal LMS [26], and JUN oncogene amplification and over-expression block adipocytic differentiation in highly aggressive sarcomas. Most frequently, LMS have appeared in the uterus, retroperitoneum or extremities, and although histologically indistinguishable, they have different clinical courses and chemotherapeutic responses. The molecular basis for these differences remains unclear, therefore, the examination of human uterine LMS tissues (23 LMS tissue sections and normal tissue sections located in the same tissue) was performed to detect somatic (tumour-specific) mutations in the IFN-γ signal molecules. In a recent report, high-resolution genome wide array comparative genomic hybrydization (CGH) analysis of LMS cases gave gene-level information about the amplified and deleted regions that may play a role in the development and progression of human U-LMS. Among the most intriguing genes, whose copy number sequence was revealed by CGH, were loss of JAK1 (1p31-p32) and PSMB9/β1i (6p21.3) [16,17]. The discovery of these mutational defects in a key cell-signaling pathway may be an important development in the pathogenesis of human U-LMS.
The growth of JAK1-deficient cell lines reportedly is unaffected; similarly, the cell cycle distribution pattern of freshly explanted tumour cells derived from JAK1-deficient tumours shows no response to IFN-γ signaling [27]. The growth of the original SKN cells, which had defective JAK1 activity, was unaffected by IFN-γ treatment. In contrast, the growth of JAK1-transfected SKN cells, which had strong exogenous JAK1 activity, was prevented by IFN-Γ treatment. Interestingly, when PSMB9/β1i-transfected SKN cells, which have marked the expression of PSMB9/β1i, were analyzed, expression of exogenous PSMB9/β1i resulted in cell growth inhibition. Conversely, the growth of PSMB9/β1i-transfected SKN cells was unaffected by IFN-Γ signal pathway. Taken together, IFN-γ response to cell growth inhibition may be attributable to the physiological significance of PSMB9/β1i.
In conclusion, the down regulation of major histocompatibility complex (MHC) expression, including the TAP1 and PSMB9/β1i genes, is one of the biological mechanisms tumour cells use to evade host immunosurveillance [28-30]. Recently, the incidence of IFN-γ unresponsiveness in human tumours was examined in several cancers, and revealed that approximately 33% of each group exhibited a reduction in IFN-γ sensitivity [31]. Nevertheless, the expression of PSMB9/β1i, rather than providing an escape from immune surveillance, seems to play an important role in the negative regulation of human U-LMS cell growth. Defective expression of PSMB9/β1i is likely to be one of the risk factors for the development of human uterine neoplasm, as it is in the PSMB9/β1i-deficient mouse. Thus, gene therapy with PSMB9/β1i expression vectors may be a new treatment for U-LMS that exhibits a defect in the expression of PSMB9/β1i. Because there is no effective therapy for unresectable U-LMS, our results may bring us to specific molecular therapies to treat this disease.
We sincerely appreciate the generous donation of PSMB9/β1i-deficient breeding mice and technical comments by Dr. Susumu Tonegawa, Massachusetts Institute of Technology. We thank Isamu Ishiwata for his generous gift of the U-LMS cell lines. This work was supported by grants from the Ministry of Education, Culture, Science and Technology, the Japan Science and Technology Agency, the Foundation for the Promotion of Cancer Research, Kanzawa Medical Research Foundation, and The Ichiro Kanehara Foundation.
- Zaloudek C, Hendrickson MR (2002) Mesenchymal tumors of the uterus, in Kurman RJ. (ed): Blaustein‘s Pathology of the Female Genital Tract (ed 5). New York, Springer-Verlag: 561-578.
- Gadducci A, Landoni F, Sartori E, Zola P, Maggino T. et al. (1996) Uterine leiomyosarcoma: analysis of treatment failures and survival. Gynecol Oncol 62: 25-32.
- Nordal RR, Thoresen SO (1997) Uterine sarcomas in Norway 1956-1992: incidence, survival and mortality. Eur J Cancer 33: 907-911. [Crossref]
- Brooks SE, Zhan M, Cote T, Baquet CR (2004) Surveillance, epidemiology, and end results analysis of 2677 cases of uterine sarcoma 1989–1999. Gynecol Oncol 93: 204-208.
- Dusenbery KE, Potish RA, Argenta PA, Judson PL (2005) On the apparent failure of adjuvant pelvic radiotherapy to improve survival for women with uterine sarcomas confined to the uterus. Am J Clin Oncol 28: 295-300.
- Wu TI, Chang TC, Hsueh S, Hsu KH, Chou HH, et al. (2006) Prognostic factors and impact of adjuvant chemotherapy for uterine leiomyosarcoma. Gynecol Oncol 100: 166-172.
- Leitao MM, Soslow RA, Nonaka D, Olshen AB, Aghajanian C. et al. (2004) Tissue microarray immunohistochemical expression of estrogen, progesterone, and androgen receptors in uterine leiomyomata and leiomyosarcoma. Cancer 101: 1455-1462.
- Perez EA, Pusztai L, Van de Vijver M (2004) Improving patient care through molecular diagnostics. Semin Oncol 31: 14-20. [Crossref]
- Miettinen M, Fetsch JF (2006) Evaluation of biological potential of smooth muscle tumours. Histopathology 48: 97-105. [Crossref]
- Bodner-Adler B, Bodner K, Czerwenka K, Kimberger O, Leodolter S, et al. (2005) Expression of p16 protein in patients with uterine smooth muscle tumors: an immunohistochemical analysis. Gynecol Oncol 96: 62-66.
- Van Kaer L, Ashton-Rickardt PG, Eichelberger M, Gaczynska M, Nagashima K, et al. (1994) Altered peptidase and viral-specific T cell response in LMP2 mutant mice. Immunity 1: 533-541. [Crossref]
- Hayashi T, Faustman DL (2002) Development of spontaneous uterine tumors in low molecular mass polypeptide-2 knockout mice. Cancer Res 62: 24-27. [Crossref]
- Hayashi T, Kobayashi Y, Kohsaka S, Sano K (2006) The mutation in the ATPbinding region of JAK1, identified in human uterine leiomyosarcomas, results in defective interferon-gamma inducibility of TAP1 and LMP2. Oncogene 25: 4016-4026.
- MOTIF Search profiling. http://motif.genome.jp
- NCBI’s Conserved Domain Database and Search Service, v2.17 analysis. http:// www.ncbi.nlm.nih.gov/ Structure/cdd/cdd.shtml
- Larramendy ML, Kaur S, Svarvar C, Bo¨hling T, Knuutila S (2006) Gene copy number profiling of soft-tissue leiomyo- sarcomas by array comparative genome hybridization. Cancer Genet Cytogen 169: 94-101.
- Svarvar C, Larramendy ML, Blomqvist C, Gentile M, Koivisto-Korander R, et al. (2006) Do DNA copy number changes differentiate uterine from nonuterine leiomyosarcomas and predict metastasis? Modern Pathol 19: 1068-1082.
- Hayashi T, Horiuchi A, Sano K, Hiraoka N, Ichimura T, et al. (2014) Potential diagnostic biomarkers: LMP2/?1i and Cyclin B1 differential expression in human uterine mesenchymal tumors. Tumori 100: 509-516.
- Kanamori T, Takakura K, Mandai M, Kariya M, Fukuhara K, et al. (2003) PEP-19 overexpression in human uterine leiomyoma. Mol Hum Reprod 9: 709-717. [Crossref]
- Wang L, Felix JC, Lee JL, Tan PY, Tourgeman DE, et al. (2003) The proto-oncogene c-kit is expressed in leiomyosarcomas of the uterus. Gynecol Oncol 90: 402-406. [Crossref]
- Ylisaukko-oja SK, Kiuru M, Lehtonen HJ, Lehtonen R, Pukkala E, et al. (2006) Analysis of fumarate hydratase mutations in a population- based series of early onset uterine leiomyosarcoma patients. Int J Cancer 119: 283-287.
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, et al. (2004) A census of human cancer genes. Nat Rev Cancer 4: 177-183. [Crossref]
- Jones PA, Baylin SB (2007) The epigenomics of cancer. Cell 128: 683-692. [Crossref]
- Lengyel E, Sawada K, Salgia R (2007) Tyrosine kinase mutations in human cancer. Curr Mol Med 7: 77-84. [Crossref]
- Pajares MJ, Ezponda T, Catena R, Calvo A, Pio R, et al. (2007) Alternative splicing: an emerging topic in molecular and clinical oncology. Lancet Oncol 8: 349-357. [Crossref]
- Post SM (2012) Mouse models of sarcomas: critical tools in our understanding of the pathology. Clinical Sarcoma Res 2: 20.
- Sexl V, Kovacic B, Piekorz R, Moriggl R, Stoiber D, et al. (2003) Jak1 deficiency leads to enhanced Abelson-induced B-cell tumor formation. Blood 101: 4937-4943. [Crossref]
- Singal DP, Ye M, Ni J, Snider DP (1996) Markedly decreased expression of TAP1 and LMP2 genes in HLA class I-deficient human tumor cell lines. Immunol Lett 50: 149-154.
- Dovhey SE, Ghosh NS, Wright KL (2000) Loss of interferon-gamma inducibility of TAP1 and LMP2 in a renal cell carcinoma cell line. Cancer Res 60: 5789-5796. [Crossref]
- Cabrera CM, Jimenez P, Cabrera T, Esparza C, Ruiz-Cabello F, et al. (2003) Total loss of MHC class I in colorectal tumors can be explained by two molecular pathways: beta2-microglobulin inactivation in MSI-positive tumors and LMP7/TAP2 downregulation in MSI-negative tumors. Tissue Antigens 61: 211-219.
- Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, et al. (1998) Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci USA 95: 7556-7561.
- Parmar S, Platanias LC (2005) Interferons. Cancer Treat Res 126: 45-68. [Crossref]
- Platanias LC (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5: 375-386. [Crossref]
- Bardelli A, Parsons DW, Silliman N, Ptak J, Szabo S, et al. (2003) Mutational analysis of the tyrosine kinome in colorectal cancers. Science 300: 949. [Crossref]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, et al. (2004) A census of human cancer genes. Nat Rev Cancer 4: 177-183. [Crossref]