Abstract
It is reported that 60% of congenital bilateral sensorineural hearing loss is caused by genetic factors, and half of hearing loss at a later stage is due to a single gene mutation. In this study; it is aimed to investigate the hearing loss molecular etiology of GJB2 gene mutations in patients with hearing loss. Forty-six patients who had 90 decibels and above-bilateral sensorineural hearing loss were included. DNAs of the patients were isolated from peripheral blood-EDTA samples. By using PCR primers, specific for the 1st and 2nd GJB2 gene regions, changes in the selected gene regions were investigated by DNA sequence analysis. When 46 patients with hearing loss (5 female, 41 male) were examined, pathological variation was found in 6 patients (13%) and any variation was found in 11 patients (24%). Mutations detected in the cases include heterozygous 35delG (a frameshift mutation in GJB2), heterozygous V153I and V27I (missense mutations in GJB2); and the homozygous H100P variation. No mutation was detected in the 1st exon of GJB2 gene. In the second exon of GJB2 gene; heterozygous 35delG mutation in 2 patients (4.3%), the heterozygous V27I mutation in 2 patients (4.3%), the heterozygous V153I mutation in 2 patients (4.3%) and homozygous H100P alteration in 33 patients (71.7%) were found. The variation we most frequently observed in cases following GJB2 gene sequencing was homozygous H100P alteration; This variation was assessed as polymorphism (71.7%). However, in order to determine if this variation might be related to hearing loss, it was planned to determine the presence of the H100P alteration in control group with complete healthy hearing.
Key words
Connexin 26 gene, DFN1B, non-syndromic hearing loss, mutation profiles
Introductıon
Hearing loss is defined as the loss of hearing originated from pathology of external, middle or inner part of ear, or from a pathology of the auditory vestibular nerve or of the auditory cortex [1]. It is the most common sensory deficit in Turkey and in the world [2]. The incidence of congenital hearing loss is estimated at about 1 or 2 out of every 1000 children in the world, and more than seventy million people are affected by hearing loss [3]. Additionally, prelingual sensorineural hearing loss is the most common sensorial disorder in children with a birth frequency of 1/650 [4].
Hearing loss affects language acquisition and the progress of education in newborns; and also seriously affects socio-emotional development and quality of lives in the period of childhood and adulthood [5]. For this reason, early detection of the causes are very important. It is now known that we noticed the role of sound recognition and transmission in our lives, more than 130 gene loci; 54 autosomal-dominant, 67 autosomal-recessive, 8 X-linked, 1 Y-linked and 2 mitochondrial genes in the human genome and perhaps even more are necessary for the hearing system [6]. Approximately, 40-60% of hearing loss are of genetic origin; nonsyndromic hearing loss accounts for 70% of genetic hearing loss. Nonsyndromic genetic prelingual sensorineural hearing loss is inherited in a recessive mode in approximately 80% of cases, in a dominant mode in approximately 20% and is either X-linked or mitochondrial origin in 2 to 3% of cases [7,8].
Various guidelines are used to identify genetic hearing loss. As a symbol for genetic nonsyndromic hearing loss, the symbol DFN, produced from “DeafNess”, is used. The prefix, “DFNA”, is used for autosomal dominant form and “DFNB” is used for autosomal recessive form. Only “DFN” symbolizes X-linked and mitochondrial inheritance [9]. As a rule of thumb, most prelingual cases with hereditary impaired hearing follow a recessive segregation, whereas most cases of postlingual hereditary impaired hearing follow an autosomal dominant trait [10-12].
Nonsyndromic hearing loss is usually monogenic but shows high degree of genetic heterogeneity. The origin of damage in inherited nonsyndromic hearing loss is mostly sensorineul. According to the National Institute on Deafness and Other Communication Disorders (NIDCD), of the three types of hearing loss, more than 90 percent is sensorineural in nature. Mutations of GJB2 gene rank the first among the genes that cause nonsyndromic sensorineural hearing loss in many populations worldwide [13-15]. 35delG, the most common GJB2 mutation, covers 48-77% of GJB2 gene mutations [16-18]. GJB2 gene was shown for the first time in two large families of Tunisia with prelingual, severe hearing loss with the help of linkage analysis [19]. This gene was first described in the DFNB1A locus in 1997. In this study, Kelsell et al. identified a homozygous mutation in GJB2 gene affecting the members of 3 families mentioned in the article. Immunohistochemical staining has shown that Cx26 is highly expressed in human cochlear cells [13].
In the current study; it was aimed to investigate the possible point mutations in GJB2 gene in patients with hearing loss in Canakkale population.
Materıals and methods
Patient recruitment
Hearing loss diagnosis was formed after with medical history, physical examination, audiological evaluation, laboratory findings and radiological imaging. In the medical story; age of the patients, how many children were present in the family, of which children have hearing loss and anomaly; if there were any other individuals who have hearing loss in the family including close relatives; severity of hearing loss, parental consanguinity were questioned. Patients who applied directly to Canakkale Onsekiz Mart University, Faculty of Medicine, Department of Medical Genetics or referred from otolaryngology specialist to us with 90 decibels and above bilateral sensorineural hearing loss started before the age of 2.5 years were included. In a total of forty-six patients who had 90 decibels and above-bilateral sensorineural hearing loss were included in the current study. DNAs of the patients were isolated from peripheral blood-EDTA samples. By using PCR primers, specific for the 1st and 2nd GJB2 gene regions, changes in the selected gene regions were investigated by DNA sequence analysis. Our project was initiated on 25.09.2013 with the permission number of EK-2013-162 taken from Çanakkale Onsekiz Mart University Ethics Committee of Clinical Investigations and supported by Çanakkale Onsekiz Mart University Scientific Research Projects Commission with the project number TYL-2014/204.
DNA isolation and genotyping
A 1-2 mL peripheral blood sample from each patient's forearm was taken into a tube containing EDTA. The blood samples were stored at -20 C for later use. DNA extraction and molecular analyses were carried out in Canakkale Onsekiz Mart University Faculty of Medicine Medical Genetics Laboratory. HighPure PCR Template Preparation Kit was used for DNA isolation according to the protocol of the manufacturer and the spin column-based purification method (Roche, Germany)
PCR
SeqFinder Squencing System Connexin (GJB2) Kit Procedure provided by GML company (GML, Switzerland) was used for amplification of the Exon 1, Exon 2A and Exon 2B regions of connexin 26 gene from genomic DNA isolated from blood samples. Suitable primer sets including the polymorphic region were used to amplify each gene. Therefore, PCR was performed using the forward and reverse primer sets specific to the region of interest. Two primers, F and R, were used for each region. As the total reaction volume was 15 μl, the components were mixed with 0.2 μl sterile tubes. Taq DNA polymerase was recently added to the prepared PCR mixture. The mixture was thoroughly homogenized. PCR was performed on an apparatus (Applied Biosystem Thermal Cycler) that provided an automatic temperature cycle. After PCR, the PCR products were subjected to ExoSAP-IT and purification steps.
Primer sequences that used for target gene amplification
Gene |
Primer |
Ekzon 1-F |
TATGTTCCTGTGTTGTGTGCAT |
Ekzon 1-R |
TTTGATCTCCTCGATGTCCTTA |
Ekzon 2-F |
GTGGCCTACCGGAGACAT |
Ekzon 2-R |
CCCTCTCATGCTGTCTATTTCTT |
Sanger direct sequencing
The products (10-12 μl) obtained after the purification process were loaded on an automatic sequence analyzer (Genetic Analyzer / ABI Prism 3130) with a capillary electrophoresis system to determine the exon 1, exon 2A and exon 2B region sequences of connexin 26 gene. Seqscape v2.6 software was used to read the device.
Results
DNA samples from 46 patients (5 Female, 41 Male) with hearing loss aged between 13-24 were subjected to PCR and the first and second exons of GJB2 gene were sequenced. Mutations were detected in 6 of the patients (13%) and no variation was found in 11 patients (24%). These mutations were heterozygous 35delG heterozygous V153I and heterozygous V27I mutations. While no mutation was detected in the first exon, various point mutations were detected in the second exon of GJB2 gene in the current cohort; two patients(4.3%) had heterozygous 35delG (Figure 1), two patients (4.3%) had heterozygous V27I mutations (Figure 2), two patients(4.3%) had heterozygous V153I mutations (Figure 3) and 33 patients (71.7%) had homozygous for H100P variation in the presented results.
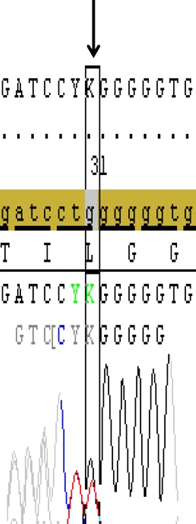
Figure 1. Sanger profiling of target gene shows the heterozygous 35delG deletion that resulted with frameshift mutation (Gly12Valfs) in the second exon of GJB2 gene in a case (Image from Seqscape v2.6 software).
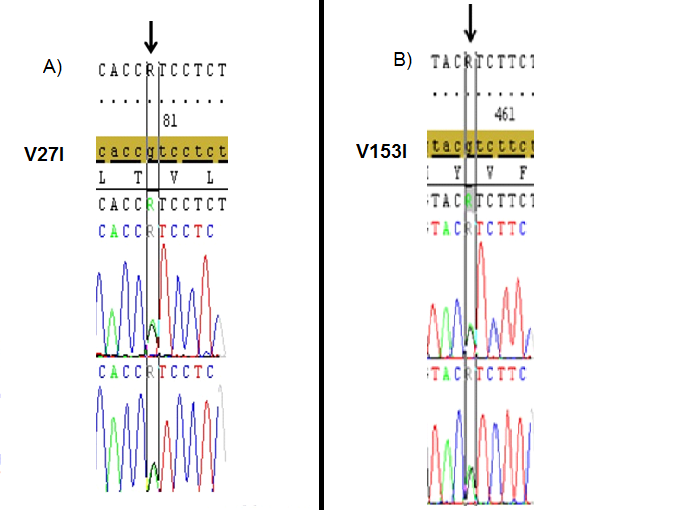
Figure 2. The heterozygous V27I (A), and V153I (B) mutations (black arrows) that identified in the 2nd exon of GJB2 gene in different two cases with hearing loss.(Image from Seqscape v2.6 software).
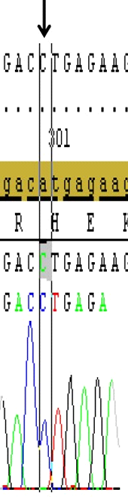
Figure 3. Shows the H100P variation (A/C transversion), (black arrow) in the second exon of the GJB2 gene that detected in most of the studied patients. (Image from Seqscape v2.6 software)
Dıscussıon
Connexins are encoded by a large gene family predicted to be composed of at least 22 isoforms [20]. This protein exhibits a non-glycosylated, structurally conserved property of 25-62 kDa in size, which differs in the length of the C-terminal domain. According to the gene constructs, gene homologues, specific sequence motifs; connexins are divided into alpha, beta and gamma groups. In addition, in the literature connexins are grouped according to the molecular masses (26 kDa = Cx26) and evolutionary classification (GJB2=Cx26). The processes involving the biosynthesis of the subunits of connexin in the endoplasmic reticulum membrane are very tightly regulated. Many processes are involved, including oligomerization of subunits appropriate for hexameric hemi-channels , insertion of connexins into the plasma membrane, head-to-head docking with partner hexameric channels positioned on neighbouring cells, regulation of dynamic, spatial, temporally organized plaques, degradation and delivery of the channels to the cytoplasm [21].
Membrane topology shared by all Cx isoforms, which includes transmembrane (membrane-spanning) domains (M1-M2-M3-M4) that are connected by two extracellular domains (extracellular loops) (ELs), one cytoplasmic domain (cytoplasmic loop-intracellular loop) (CL-IL), one N-terminal (amino) domain (NT) and one C-terminal (carboxi) domain (CT) of each hemichannel face the cytoplasm. The length of the NT and CT segments is not intended to represent any particular Cx isoform [22,23]. The N-terminal domain plays a role in voltage change by making covalent and non-covalent modifications in an environment surrounded by aminocytes. Transmembrane domains provide the formation of gap junction channel pores and channel permeability.The M1 domain operates as a voltage sensor. The M2 domain is required for oligomerization of the connexins. The M3 domain exhibits amphipathic properties, which are necessary for channel discrimination [22]. There are 5 isoforms of connexin in the mature mammalian cochlea. These are; Cx26, Cx29, Cx30, Cx31 and Cx43 [24]. Recent findings show that the connexins have single and specific functions. Although each isoform of connexin provides different tissue distribution, many cell types generate more than one isoform. When investigating the oligomerisation behavior of different isoforms of connexin, it is seen that not all of the connexins participate in the heterooligomeric connexin form. These observations show that connexin isoform interaction are selective. Although we do not know why many different channel types are present, it is reported that it may be necessary to preserve many different specific needs of the various cells to which they are expressed [25]. GJB2 gene mutations cause the potassium cycle and the endococlear potential to deteriorate, leading to hearing loss. The reason of this is the inhibition of receptor potential in hair cells that activate afferent nerve fibers. Mutations affecting GJB2 gene cause disruption of the potassium cycle resulting in local intoxication of the organ of corti by potassium, causing cell death and consequently hearing loss. It is not known exactly which areas are damaged due to the Cx 26 mutation along the auditory pathway in cochlea [26]. One of the most common CX26 mutations in humans is 35delG and is responsible for over half of all pathological Cx26 mutations [18]. The mutation appears as a result of a single nucleotide deletion in the chain consisting of 6 guanine residues starting at position 30 and ending at position 35. This results in premature termination of the 12th amino acid chain [13,27-30].
The emergence of this mutation was based on an age of about 500 generations ago and was determined to be 1000 years old. The mutation probably originated in some parts of the Middle East, followed two neolithic migration routes and spread all over Europe [29,31]. 35delG mutation, with an old deletion mutation spreading to Europe and the Middle East, has completely independent and repetitive features. Turkey, which acts as a bridge between Asia and Europe, is situated on two mutation migration routes. Even if 35delG is an ancestral mutation and a frequent mutation among the populations, there should be a region / regions and an ancestral haplotype / haplotypes where our population participates in the gene pool [32].
In white race, 35d2021 Copyright OAT. All rights reserv a 'hot spot' region. It is reported that six G repeats between nucleotides 30 and 35 of the second exon are an error point for DNA polymerase alpha and cause to be mutational hot spots in it. However, other studies using SNP markers have shown that; 35delG is a 'founder effect' mutation, with which it's frequency increased in the population, rather than 'hot spot' mutation. The selective association of hearing loss individuals with each other causes a shift in the frequencies of these pathological alleles in the gene pool. This mutation causes glycine amino acid to be converted to valine amino acid in codon 12, leading to the formation of a premature stop codon. Clinical phenotypes of those who are homozygous for this mutation can range from moderate hearing loss to very severe hearing loss [5].In different studies about the incidence of 35delG carrier in Turkey, 0.8%, 1.8% and 2.7% results were found [32-35]. Various studies have indicated that 63-79% of nonsyndromic recessive hearing loss in Mediterranean and Turkish populations is due to this mutation disorder [30,36].
One of the most comprehensive studies on the etiology of deafness in Turkey is the pedigree and clinical analysis of 840 deaf students. In this study, it was reported that 51% of the deaf probands were of genetic origin, 15% had subsequently acquired factors, and 34% were not known the cause of deafness [37]. In the study of Silan et al. in 2010; all of 64 students in Gallipoli Hearing Impairment School had had clinical assessments, pedigree analyzes, and 35delG and Mit A1555G mutation analysis; 40 cases of them were diagnosed as genetic origin (62.5%), 10 cases were defined as genetic and acquired etiology (15.6%), 13 cases were diagnosed as acquired origin (20.3%) and only 1 case (1.6%) was defined as without etiology [38].
In our study; in order to base the disease on genetic causes, the detailed family history from each family and the fact that more than one deaf individual in the family have provided supportive results for hereditary hearing loss.Individuals in this study with heterozygous 35delG mutation show hearing loss at advanced levels. In these cases, there may be mutations in the intronic region of connexin 26 gene or in other connexin genes. Intronic mutations may also interfere with normal polypeptide synthesis by disrupting splicing.
In Mediterranean populations, carrier frequency of 35delG was assessed at around 1 in 49 overall [39]. A prominently higher carrier frequency of the mutation 35delG was identified, with 3.5% and 4% in Greek and Italian populations, respectively [33,40]. In terms of 35delG mutation, it was reported that Turkey did not show a homogenous structure, it was a significant difference between western and eastern part of Turkey and that the frequency of homozygous 35delG was lower in the East part of Turkey although consanguineous marriage rate was high [5]. In our study, the incidence of 35delG in 46 probands included in was found to be 4.3%. The V27I mutation in GJB2 gene is the result of adenine replacement in place of guanine at nucleotide 79 position and conversion of valine to isoleucine at codon position 27 in the first transmembrane region. It's frequency is below 1% in European populations [41]. In our findings, the rate of occurrence in 46 patients was 4.3%.The variation of V153I was considered as polymorphism in some studies and mutation in others. For example, in a study conducted with the DHPLC technique, a heterozygous mutation in a patient was detected in the study in which this variation was considered to be as mutation like in our study. Severe hearing loss associated with radiological findings such as aplasia in the cochlear nerve and narrowing of the internal auditory canal of the patient has been reported [5,42,43]. In another study conducted in 14 different families of our country, only 2 patients were found and evaluated as polymorphism [44]. In our study, the V153I mutation was found heterozygous in two of the probands, with a 4.3% incidence overall.
The most frequent variation in our study, homozygous H100P variation, was assessed as polymorphism and found in 33 hearing impaired patients (71.7%). In a study by Snoeckx and colleagues in a large multicenter study of GJB2 mutations and the degree of hearing loss, the frequency of H100P variation was significantly lower among patients included in the study [41]. Moreover, Gardner et al. in 2006 found that in the study they performed, the H100P variation was very rare [43]. Therefore, it is not wrong to evaluate the H100P variation as polymorphism. While homozygous H100P variation was detected in 7 of 9 hearing-impaired siblings, heterozygous 35delG mutation and homozygous H100P variation detection in one patient suggested that mutations could have a combined effect. In addition to hearing loss, this patient also had spinal dysraphism findings. However, in order to determine whether H100P is associated with hearing loss, comparison of the incidence of H100P variation in healthy hearing controls should be considered in further studies.
Acknowledgements
Authors want to thank to scientific research and project supporting unit- BAP for their kindly funding of the current results (Grant no: TYL-204/2014), Canakkale Onsekiz Mart University, Canakkale/Turkey.
References
- Isaacson JE, Vora NM (2003) Differential diagnosis and treatment of hearing loss. Am Fam Physician 68: 1125-1132. [Crossref]
- Hilgert N, Huentelman MJ, Thorburn AQ, Fransen E, Dieltjens N, et al. (2009) Phenotypic variability of patients homozygous for the GJB2 mutation 35delG cannot be explained by the influence of one major modifier gene. Eur J Hum Genet 17: 517-524. [Crossref]
- Morton CC, Nance WE (2006) Newborn hearing screening—a silent revolution. N Engl J Med 354: 2151-2164. [Crossref]
- Tamayo ML, Olarte M, Gelvez N, Gómez M, Frías JL, et al. (2009) Molecular studies in the GJB2 gene (Cx26) among a deaf population from Bogota, Colombia: results of a screening program. Int J Pediatr Otorhinolaryngol 73: 97-101. [Crossref]
- Eyerci N (2010) Evaluation of mutations in the Connexin 26(GJB2) gene causing genetic hearing deficiency by Microarray-Nanochip method; regional investigation. Ataturk Univ Fac Med Dep Med Biol: 122.
- Lingala HB, Sankarathi, Penagaluru PR (2009) Role of connexin 26 (GJB2) & mitochondrial small ribosomal RNA (mt 12S rRNA) genes in sporadic & aminoglycoside-induced non syndromic hearing impairment. Indian J Med Res 130: 369-378. [Crossref]
- Morton NE (1991) Genetic epidemiology of hearing impairment. Ann N Y Acad Sci 630: 16-31. [Crossref]
- Brownstein Z, Bhonker Y, Avraham KB (2012) High-throughput sequencing to decipher the genetic heterogeneity of deafness. Genome Biol 13: 245.
- Lalwani AK, Castelein CM (1999) Cracking the auditory genetic code: nonsyndromic hereditary hearing impairment. Am J Otol 20: 115-132. [Crossref]
- Bitner-Glindzicz M (2002) Hereditary deafness and phenotyping in humans. Br Med Bull 63: 73-94. [Crossref]
- Smith RJH, Hone S (2003) Genetic screening for deafness. Pediatr Clin North Am 50: 315-329. [Crossref]
- Petersen M (2002). Non‐syndromic autosomal‐dominant deafness. Clin Genet 62: 1-13. [Crossref]
- Kelsell DP, Dunlop J, Stevens HP, Lench NJ (1997) Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 387: 80-83.
- Van Camp G, Willems PJ, Smith RJ (1997) Nonsyndromic hearing impairment: unparalleled heterogeneity. Am J Hum Genet 60: 758-764. [Crossref]
- Gasparini P, Estivill X, Volpini V, Totaro A, Castellvi-Bel S, et al. (1996) Linkage of DFNB1 to non-syndromic neurosensory autosomal-recessive deafness in Mediterranean families. Eur J Hum Genet EJHG 5: 83-88. [Crossref]
- Kalay E, Caylan R, Kremer H, de Brouwer APM, Karaguzel A (2005) GJB2 mutations in Turkish patients with ARNSHL: prevalence and two novel mutations. Hear Res 203: 88-93. [Crossref]
- Hayashi C, Funayama M, Li Y, Kamiya K, Kawano A, et al. (2011) Prevalence of GJB2 causing recessive profound non-syndromic deafness in Japanese children. Int J Pediatr Otorhinolaryngol 75: 211-214.
- Cohn ES, Kelley PM (1999) Clinical phenotype and mutations in connexin 26 (DFNB1/GJB2), the most common cause of childhood hearing loss. Am J Med Genet Part A 89: 130-136. [Crossref]
- Guilford P, Arab S Ben, Blanchard S, Levilliers J, Weissenbach J, Belkahia A, et al. (1994) A non–syndromic form of neurosensory, recessive deafness maps to the pericentromeric region of chromosome 13q. Nat Genet 6: 24-28. [Crossref]
- Thönnissen E, Rabionet R, Arbonès M, Estivill X, Willecke K, et al. (2002) Human connexin26 (GJB2) deafness mutations affect the function of gap junction channels at different levels of protein expression. Hum Genet 111: 190-197. [Crossref]
- Evans WH, De Vuyst E, Leybaert L (2006) The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem J 397: 1-14. [Crossref]
- Krutovskikh V, Yamasaki H (2000) Connexin gene mutations in human genetic diseases. Mutat Res Mutat Res 462: 197-207. [Crossref]
- Retamal MA, Reyes EP, García IE, Pinto B, Martínez AD, et al. (2015) Diseases associated with leaky hemichannels. Front Cell Neurosci 9: 267. [Crossref]
- Forge A, Marziano NK, Casalotti SO, Becker DL, Jagger D (2003) The inner ear contains heteromeric channels composed of cx26 and cx30 and deafness-related mutations in cx26 have a dominant negative effect on cx30. Cell Commun Adhes 10: 341-346. [Crossref]
- Bruzzone R, White TW (1996) Goodenough DA. The cellular internet: On‐line with connexins. Bioessays 18: 709-718. [Crossref]
- Martínez AD, Acuña R, Figueroa V, Maripillan J, Nicholson B (2009) Gap-junction channels dysfunction in deafness and hearing loss. Antioxid Redox Signal 11: 309-322. [Crossref]
- Reardon W (1998) Connexin 26 gene mutation and autosomal recessive deafness. Lancet 351: 383-384. [Crossref]
- Park H, Houn Hahn S, Chun Y, Park K, Kim H (2000) Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope 110: 1535-1538. [Crossref]
- Laer L Van, Camp G Van (2001) Genes in the ear: what have we learned over the last years? Scand Audiol Suppl 30: 44-53. [Crossref]
- Denoyelle F, Marlin S, Weil D, Moatti L, Chauvin P, et al. (1999) Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin-26 gene defect: implications for genetic counselling. Lancet 353: 1298-1303. [Crossref]
- Van Laer L, Coucke P, Mueller RF, Caethoven G, Flothmann K, et al. A common founder for the 35delG GJB2gene mutation in connexin 26 hearing impairment. J Med Genet 38: 515-518. [Crossref]
- Barış I, Kılınç MO, Tolun A (2001) Frequency of the 35delG mutation in the connexin 26 gene in Turkish hearing‐impaired patients. Clin Genet 60: 452-455. [Crossref]
- Gasparini P, Rabionet R, Barbujani G, Melchionda S, Petersen M, et al. (2000) High carrier frequency of the 35delG deafness mutation in European populations. Eur J Hum Genet EJHG 8: 19-23. [Crossref]
- Tekin M, Akar N, Cin Ş, Blanton S, Xia X, et al. (2001) Connexin 26 (GJB2) mutations in the Turkish population: implications for the origin and high frequency of the 35delG mutation in Caucasians. Hum Genet 108: 385-389.
- Uyguner O, Emiroglu M, Uzumcu A, Hafiz G, Ghanbari A, et al. (2003) Frequencies of gap‐and tight‐junction mutations in Turkish families with autosomal‐recessive non‐syndromic hearing loss. Clin Genet 64: 65-69. [Crossref]
- Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, et al. (1998) Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet 351: 394-398. [Crossref]
- Ozturk O, Silan F, Oghan F, Egeli E, Belli S, et al. (2005) Evaluation of deaf children in a large series in Turkey. Int J Pediatr Otorhinolaryngol 69: 367-373. [Crossref]
- Silan F, Guclu O, Kadioglu LE, Silan C, Atik S, et al. (2011) GJB2 35delG and Mitochondrial A1555G Mutations and Etiology of Deafness at the Gelibolu School for the Deaf in Turkey. Int Adv Otol 7: 361-371.
- Lucotte G, Mercier G (2001) Meta-analysis of GJB2 mutation 35delG frequencies in Europe. Genet Test 5: 149-152. [Crossref]
- Antoniadi T, Rabionet R, Kroupis C, Aperis GA, Economides J, et al. (1999) High prevalence in the Greek population of the 35delG mutation in the connexin 26 gene causing prelingual deafness. Clin Genet 55: 381-382. [Crossref]
- Snoeckx RL, Huygen PLM, Feldmann D, Marlin S, Denoyelle F, et al. (2005) GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet 77: 945-957. [Crossref]
- Guerci VI, Grasso DL, Morgutti M, Amoroso A, D’Andrea P, et al. (2007) Connexin 26 gene: Defining the role of the V1531 mutation. Audiol Med 5: 200-206.
- Gardner P, Oitmaa E, Messner A, Hoefsloot L, Metspalu A, et al. (2006) Simultaneous multigene mutation detection in patients with sensorineural hearing loss through a novel diagnostic microarray: a new approach for newborn screening follow-up. Pediatrics 118: 985-994. [Crossref]
- Bayazıt YA, Cable BB, Cataloluk O, Kara C, Chamberlin P, et al. (2003) GJB2 gene mutations causing familial hereditary deafness in Turkey. Int J Pediatr Otorhinolaryngol 67: 1331-1335. [Crossref]