Abstract
The 1990 Hannover Bone Marrow Classification separated Ph1+ CML from the Ph1- myeloproliferative neoplasms (MPN) essential thrombocythemia (ET), polycythemiavera (PV) and chronic or primary megakaryocytic granulocytic myeloproliferation (CMGM/PMGM). The 2000-2008 European Clinical Molecular and Pathological (ECMP) criteria discovered 3 variants of thrombocythemia: ET with features of PV (prodromal PV) versus “true” normocellular ET and hypercellular ET associated with CMGM/PMGM without features of PV, MDS or CML. The 2008 World Health Organization (WHO)-ECMP and 2007-2015 WHO-ECMP MPN classifications defined three phenotypes of JAK2V617F mutated ET: normocellular ET (WHO-ET), hypercelluar ET due to increased erythropoiesis (prodromal PV) and ET with hypercellular megakaryocytic-granulocytic myeloproliferation (ET.MGM) or masked PV. JAK2 wild type MPL515 mutated ET is the second distinct thrombocythemia featured by clustered giant megakaryocytes with hyperlobulated stag-horn-like nuclei, in a normocellular bone marrow consistent with the diagnosis of “true” ET. JAK2/MPL wild type hypercellular ET is the third distinct thrombocythemia characterized by clustered larged immature dysmorphic megakaryocytes and bulky (bulbous) hyperchromatic nuclei consistent with CMGM/PMGM.
Key words
myeloproliferative neoplasms, essential thrombocythemia, polycythemia vera, primary megakaryocytic granulocytic myeloproliferation, myelofibrosis, JAK2V617F mutation, MPL515 mutation, bone marrow pathology
Key message
The 2015 WHO-CMP criteria define three phenotypes of JAK2V617F mutated Myeloproliferative Neoplasms (MPN) essential thrombocythemia (ET), prodromal polycythemia vera (PV), hypercellular ET with megakaryocytic-granulocytic myeloproliferation (masked PV) and classical PV. JAK2 wild type MPL515 mutated normocellular ET is the second distinct thrombocythemia featured by clustered giant megakaryocytes with hyperlobulated stag-horn-like nuclei. JAK2/MPL wild type CALR mutated hypercellular ET is the third distinct thrombocythemia characterized by clustered larged immature dysmorphic megakaryocytes and bulky (bulbous) hyperchromatic nuclei consistent with CMGM/PMGM. MPN disease burden is best reflected by the degree of anemia and splenomegaly on top of mutation allele burden, bone marrow cellularity and increase of reticulin fibrosis.
Introduction
In this historical appraisal of the myeloproliferative disorders (MPD) the 1980 Rotterdam, the 1990 Hannover and versus the 1975 PVSG and European clinical and bone marrow (EuroCMP) criteria myeloproliferative disorders (MPD, Tables 1-6) are compared against the 2007-2015 WHO clinical Molecular Pathological (WHO-CMP) criteria for the diagnosis Myeloproliferative Neoplasms (MPN) and the detection and staging of the masked and manifest MPNs ET, PV and MF (Tables 7-11).
Table 1. PVSG criteria for PV [10] and diagnostic differentiation of PV from all variants of primary and secondary erythrocytoses by bone marrow histology [17].
The diagnostic criteria for polycythemia vera proposed by the polycythemia vera study group (PVSG) |
A1 |
Raised red cell mass |
B1 |
Thrombocytosis; platelets > 400 x 109/L |
Male > 36mL/kg |
Female > 32mL/kg |
A2 |
Normal arterial oxygen saturation > 92% |
B2 |
Leucocytosis > 12 × 109/L and no fever or infection. |
A3 |
Splenomegaly on palpation |
B3 |
Raised neutrophil alkaline phosphate score > 100 or raised B12 ( > 900 ng/L) or raised unsaturated B12 binding capacity ( > 2200 ng/L) |
Diagnostic differentiation of benign erythrocytes from myeloproliferative polycythemia vera by histopathology from bone marrow biopsy sections. |
Raised cell mass, RCM |
Normal RCM = apparent erythrocytosis does not exclude MPD! |
Raised RCM = absolute erythrocytosis |
Increase clustering of enlarged megakaryocytes with hyperploid nuclei is a diagnostic clue to polycythemia vera. |
Bone marrow histopathology differentiates between polycythemia vera (PV) and benign erythrocytosis. |
Benign erythrocytes in its purity |
Size, morphology and distribution of megakaryocytes are normal |
Classification of erythrocytosis: |
Primary: Truncated EPO receptor |
Disruption oxygen homeostasis in Chuvash polycythemia |
High oxygen affinity hemoglobinopathy |
Congenital autonomous EPO production? |
Acquired erythrocytosis: |
Secondary: EPO accumulation in renal disease |
Autonomous EPO production by tumor cells |
Hypoxia |
Idiopathic erythrocytosis |
Note: RCM does not distinguish between PV and primary or secondary erythrocytosis.
RCM does not distinguish between PV and inapparent PV with splenomegaly in splanchnic vein thrombosis (IPV, table 3). In IPV RCM is increased due to splenomegaly with absence of hypervolumemic symptoms. Bone marrow histology distinguishes PV from all variants of erythrocytosis with a sensitivity and specificity of 100%.
Table 2. Grading of bone marrow biopsy content of reticulin fibrosis (RF) according to Ellis et al., [41], and Wilkins et al., [171] and grading of RF, reticulin clloagen fibrosis (RCF) and myelofibrosis (MF) according to Georgii et al., (Hannover Bone Marrow Classification) [35,36] and Thiele [98-101].
Grading reticulin fibrosis (RF) [41]. |
Grading MF 2008 WHO [91]. |
Description of reticulin fibers (RF) and reticulin/collagen fibers (RCF) in myelofibrosis (MF) as a secondary event in myeloproliferative neoplasms (MPN). |
Normal RF-0. |
N MF 0. |
No reticulin fibers, occasional individual fibers or focal areas with tiny amount of reticulin fiber network. |
Slight increase RF 1. |
+ MF 0. |
Fine reticulin fiber network throughout much of section and no course reticukin fibers. |
Moderate increase RF 2. |
+ + MF 1. |
Diffuse fine reticuline network with focal collections of thick course reticulin fibers and no collagenisation. |
Marked increase
RF 3. |
+++ RCF = MF 2. |
Diffuse and dense increase in reticulin with extensive intersections, and presence of collagen fibers and no or minor osteosclerosis. |
OS Dry tap
RF 4. |
Sclerotic
RCF & O = MF 3. |
Diffuse and dense reticulin with with coarse bundles of collagen associated with significant osteosclerosis (O). |
Table 3. Comparison of clinical and laboratory features between PVSG defined PV (Group A) and Inapparent PV (IPV) (Group B) [44].
Clinical feature |
Group A PV |
Group B IPV |
P-value |
Number of caes |
85 |
18 |
|
Age (range) |
61 (27-83) |
52 (28-82) |
ns |
Sex male/female |
56/42% |
39/61% |
ns |
Splenomegaly |
44 (52%) |
15 (83%) |
< 0.005 |
Leukocytes > 12 ×10/L |
31 (36%) |
5 (28%) |
ns |
Platelets > 500 ×10/L |
40 (47%) |
10 (56%) |
ns |
Red Cell Counts x 10/L: males |
6.2 (4.9-7.4) |
5.2 (4.7-5.9) |
< 0.0002 |
Red Cell Mass males |
48.2 (36-60) |
43.3 (41-61) |
ns |
Red Cell Counts females |
6 (4.2-7.3) |
4.7 (3.7-5.5) |
< 0.003 |
Re Cell Mass females |
40.1 (32-59) |
37.3 (34-46 |
ns |
Plasma volume PV vs IPV: |
|
|
|
Increase/theoretical norm (%) |
+10 (-11,+61) |
+36 (+20, +98) |
< 0.00001 |
Conclusion: IPV is featured by normal values of haemoglobin, haematocrit and erythrocyte counts, in the absence of hypovolemic symptoms and in IPV red cell mass (RCM) is increased related to the degree of increased spleen size.
Table 4. The 1980 Rotterdam Clinical and Pathological (RCP) criteria for Essential Thrombocythemia (ET) and Polycythemia Vera (PV).
The 1980 RCP major (A) and confirmative (B) criteria for prefibrotic ET. |
A1 |
Persistent platelet count in excess of 400 × 109/L. |
A2 |
Increase and clustering of enlarged megakaryocytes in bone marrow biopsy. |
A3 |
No or slight increase of reticulin fibers (RF 0 or RF 1). |
B1 |
Presence of large platelets in a peripheral blood smear. |
B2 |
No splenomegaly (< 12 cm) or slight splenomegaly on scan (< 15 cm). |
B3 |
Increase of LAP-score and no signs of fever or inflammation. |
B4 |
Absence of Ph+ chromosome or any other cytogenetic abnormality in nucleated blood or bone marrow cells. |
The 1980 RCP major (A) and minor (B) criteria for prefibrotic PV. |
A1 |
Raised red cell mass. Male > 36 ml/kg, female > 32 ml/kg is consistent with erythrocyte count of > 6 × 1012/L [1-3]. |
A2 |
Slight, moderate or marked increase in bone marrow biopsy of clustered, enlarged pleomorphic megakaryocytes with hyperlobulated nuclei and moderate to marked increase cellularity of megakaryopoiesis/erythropoiesis or typically trilinear erythrocytic, megakaryocytic and granulopocytic myeloproliferation (EMGM). A typical PV bone marrow excludes erythrocytosis with a sensitivity and specificity of 100%. No or presence of reticuline fibers and no collagen fibers (no dry tap). |
B1 |
Thrombocythemia, persistant increase of platelet > 400 ×109/L. |
B2 |
Leukocytosis, leucocyte count > 109/L and low erythrocyte sedimentation rate. |
B3 |
Raised leukocyte alkaline phosphatase (LAP) score > 100, absence of fever or infection. |
B4 |
Splenomegaly on palpation or on isotope/ultrasound scanning. |
A1 + A2 |
Established erythrocythemic PV and one of B establishes classical PV. |
| |
|
|
Table 5A. The European Clinical and Pathological (ECP) criteria for True normocellular essential thrombocythemia (“True” ET). Source Michiels & Thiele in 2002 [91].
Clinical Criteria |
Pathological Criteria |
A1 |
Persistent increase of platelet count: grade I: 400-1500, grade II: > 1500 × 109/L. |
B1 |
Replace as follows:
Proliferation of mature large to giant megakaryocytes with hyperlobulated stagorn-like nuclei and mature cytoplasms |
A2 |
Normal spleen or only minor splenomegaly on echogram. |
A3 |
Normal LAP score, normal ESR, and increased MPV. |
B2 |
No proliferation or immaturity of granulopoiesis or erythropoiesis. |
A4 |
Spontaneous megakaryocyte colony formation (CFU-Meg). |
A5 |
No signs or cause of RT. |
B3 |
No or only borderline increase inreticulin. |
A6 |
No preceding or allied other subtype of MPD, CML, or MDS. |
A7 |
Absence of the Philadelphia chromosome. |
Table 5B. The European clinical and Pathological (ECP) criteria for prefibrotic chronic idiopathic myelofibrosis (CIMF) or primary megakaryocytic granulocytic myeloproliferation (PMGM) [91].
Updated clinicopathological criteria for the diagnosis of idiopathic myelofibrosis (IMF) or Agnogenic myeloid metaplasisa (AMM)* Source Michiels & Thiele 2002 [91]. |
Clinical Criteria |
Pathological Criteria |
A1 |
No preceding or allied other subtype of MPD, CML, OR MDS |
B1 |
Megakaryocytic and granulocytic myeloproliferation and relative reduction of erythroid precursors. Abnormal clustering and increase in atypical giant to medium- sized megakaryocytes contacting bulbulous (cloud-like) hypolobulated nuclei and definitive maturation defects. |
A2 |
Early clinical stages
Normal hemoglobin or anemia, grade I: hemoglobin ≥ 12 g/dL.
Slight or moderate splenomegaly on palpation or > 11cm on ultrasound scan or CT Thrombocythemia, platelets > 400 × 109/L. |
A3 |
Intermediate clinical stage
Anemia grade II: hemoglobin ≥ 10 g/dL.
Definitive leuko-erythroblastic blood picture and/ or tear-drop erythrocytes.
Splenomegaly.
No adverse signst. |
A4 |
Advanced clinical stage
Anemia grade III: hemoglobin < 10 g/L.
One or more adverse signst. |
Table 6. The 2000-2005 European Clinical and Pathological (ECP) Essential Thrombocythemia (ET) [77].
Clinical Criteria |
Pathological Criteria |
A1 |
Persistent increase of platelet count: in excessof 400 × 109/L. |
B1 |
Predominant proliferation of dispersed or loosely clustered enlarged to giant |
A2 |
Normal spleen or only minor splenomegaly on echogram. |
A3 |
Normal or increased LAP-score and normal ESR. |
B2 |
No or only borderline increase in reticulin. |
A4 |
Spontaneous megakaryocyte colont formaton (CFU-Meg) and or endogenous erythroid colony formation (EEC). |
A5 |
No signs or cause of reactive thrombocytosis (RTh). |
A6 |
No preceding or allied other subtype of MPD, CML OR MDS. |
A7 |
Absence of the Ph-Chromosme. |
A1 and B1 + B2 establish (true) ET. Any other additional A criterion confirms ET.
ET: GRADE I: platelet counts between 400 and 1,000 × 109/L; Grade II: platelet counts 1,000-1,500 × 109/L; and Grade III: platelet counts above 1,000 × 109/L.
Table 7. The 2000-2005 European Clinical and Pathological (ECP) criteria for polycythemia vera (PV) [77].
Clinical Criteria |
Pathological Criteria |
A1 |
Erythrocyte > 6 × 1012/L, haemoglobin: male > 18.5 g/dl, female > 16.5 g/dl, and haematocrit: male > 0.51%, female > 0.48%. Raised red cell mass (optional) RCM: male > 36 ml/kg, female > 32ml/kg. |
B1 |
Increased cellularity with trilineage myeloproliferation (i.e. panmyelosis). Proliferation and clustering of small to giant (pleomorphic) megakaryocytes. Absence of stainable iron. No pronounced inflammatory reaction (plasmacytosis, cellular debris). |
A2 |
Persistent increase of platelet count: grade I: 400-1,500, grade II: > 1500. |
A3 |
Splenomegaly on palpation or on ultrasound or CT (>12cm length diameter). |
B2 |
Spontaneous erythroid colony formation. |
A4 |
Granulocytes > 10 × 109/l and/or raised LAP-score in the absence of fever or infection. |
MF 0 |
Prefibrotic stage PV. |
MF 1 |
Early Prefibrotic stage PV. |
A5 |
Absence of any cause of secondary erythrocytosis. |
MF 2 |
Manifest myelofibrosis in PV. |
|
|
MF 3 |
Advanced myelofibrosis in PV. |
A6 |
Low plasma EPO level. |
MF > 3 |
Osteosclerosis, decreased cellularity. |
A1 + B1 + B2 establishes polycythemia vera (PV) –any other criterion confirms PV.
Table 8. 2007 EuroCMP and 2015 WHO-CMP criteria for diagnosis of JAK2V617F mutated Polycythemia Vera [95,96,1-3,104].
Clinical and molecular criteria |
Pathological criteria (WHO) |
Major PV criteria |
P1 |
Bone marrow pathology: increased cellularity due to trilinear increase of erythropoiesis, megakaryopoiesis and granulopoiesis and clustering of small to giant (pleomorph) megakaryocytes with hyperlobulated nuclei. |
A0 |
Early PV. Hematocrit in the upper limit of normal: Ht: 0.45 to 0.51 in male and 0.43 to 0.48 in female, erythrocytes < 6 × 1012/L. |
|
A1 |
Classical WHO defined PV: Hematocrit > 0.51/ > 0.48 in male/female.
Erythrocyte > 6 × 1012/L. |
|
A2 |
Presence of JAK2V617F or exon 12 mutation (Sensitivity 98%). |
|
Absence of stainable iron. No pronounced inflammatory reaction (plasmacytosis, cellular debris). |
A3 |
Low serum EPO level and/or spontaneous endogenous erythroid colony (EEC) formation. |
Minor MPN criteria |
P2 |
Selective increase of erythropoiesis, normal granulopoiesis and megakaryocytes of normal size, morphology and no clustering of megakaryocytes in primary or secondary erythrocytosis. |
B1 |
Persistent increase of platelet count grade I: 400-1500, grade II: > 1500. |
B2 |
Granulocytes > 10 × 109/L or raised LAP-score or increased PRV-1 expression in the absence of fever or infection. |
|
Splenomegaly on palpation or on ultrasound echogram ( > 12cm length in diameter). |
P3 |
Grading of reticulin fibrosis (RF) post PV-MF-2 and MF-3. |
Table 9. 2007 EuroCMP and 2015 WHO-CMP criteria for diagnosis of 3 phenotypes of JAK2V617F mutated Essential Thrombocythemia (ET) [95.96,103,104].
Clinical and molecular criteria |
WHO bone marrow criteria |
ET stage 1 |
ET |
1 |
Platelet count of > 350 × 109/l and the presence of large platelets in a blood smear (also stage 2 and 3). |
Predominant proliferation of enlarged pleiomorphic megakaryocytes with hyperlobulated nuclei and mature cytoplasm, lacking conspicuous morphological abnormalities. No increase, proliferation or immaturity of granulopoiesis or erythropoiesis. No or borderline increase in reticulin. RFO-1 |
2 |
Presence of JAK2-V617F mutation. |
3 |
Normal haematocrit: male <51%, female < 48% erythrocytes < 6 × 1012/L. |
ET stage 2 |
Prodromal PV |
1 |
Platelet count of > 350 × 109/l and haematocrit in normal or upper normal range: male <51%, female < 48, erythrocytes < 6 × 1012/l |
Increased cellularity with trilineage myeloproliferation (i.e.,panmyelosis). Proliferation and clustering of small to giant (pleomorphic) megakaryocytes. No pronounced inflammatory reaction (plasmacytosis, cellular debris). Absence bone features consistent with congenital polycythemia and secondary erythrocytosis.
No borderline increase in reticulin. RF-01 |
2 |
Presence of JAK2-V617F mutation. |
3 |
Low serum EPO level and/or increased LAP score. |
4 |
Spontaneous EEC. |
ET stage 3 |
ET MGM |
1 |
Platelet count of > 350 × 109/l and no signs of leuko-erythroblastosis. |
Increased cellularity due to megakaryocytic granulocytic myeloproliferation (MGM).
Normal or reduced erythropoiesis.
Loose to dense clustering of enlared pleiomorphic megakaryocytes with hyperploid nuclei and the presence of pleiomorphic megakaryocytes with clumsy dysmorphic lobulated nuclei.
No or borederline increase in reticulin RF0-1 |
2 |
No or slight splenomegay on ultrasound. |
3 |
No anemia with Hb and Ht in the normal or lower range of normal: > 12g/dl. |
4 |
Presence of JAK2-V617F mutation. |
5 |
No proceeding or allied of CML, PV, RARS-T OR MDS. |
Table 10. 2007 EuroCMP and 2015 WHO-CMP criteria for the diagnosis of JAK2 wild type normocellular ET carrying the MPL515 mutation (+) [95.96,103,104].
Clinical and molecular criteria |
WHO bone marrow criteria |
ET |
ET |
- Platelet count > 350-400 × 109/l. Presence of large platelets in blood smear.
|
Predominant proliferation of small, large and giant megakaryocyte with hyperlobulated, staghorn- like nuclei lacking morphological abnormalities.
No increase, proliferation or immaturity of granulopoiesis or erythropoiesis.
No or slight increase in reticulin: RF 0-1. |
- JAK-2 wild type +/- MPL515 mutation.
|
- No anemia, Hb > 12.8 g/dl.
|
ET-MF |
- Normal serum EPO and ferritin.
|
- No or slight splenomegaly on ultrasound.
|
Increased cellularity due to chronic megakaryocytic and granulocytic myeloproliferation and reduced erythroid precursors.
Loose to dense clustering of small, large and giant megakaryocytes and the presence of some dysmature clumpsy lobulated nuclei.
Increase in reticulin: RF 2,3 or 4. |
- No leukoerythroblastosis.
|
- No dry tap on bone marrow biopsy.
|
- No preceding or allied of CML, PV, RARS-T or MDS.
|
Table 11. 2007 EuroCMP and 2015 WHO-CMP criteria for hyper cellular ET associated with JAJK2 wild type CALR mutated primary megakaryocytic granulocytic myeloproliferation (PMGM) [77,78,94-96,103,104].
European clinical molecular and pathological (EuroCMP.) criteria for the diagnosis and staging of PMF or PMGM. |
J.J.Michiels |
Clinical criteria 2005 |
J.Thiele |
Pathological criteria 2005/2008 |
A1 |
No preceding allied other subtype of myeloproliferative neoplasms, CML or MDS. Main presenting feature is pronounced thrombocythemia and no dry tap on bone marrow aspiration.
JAK2 and MPL wild type. |
B1 |
Primary megakaryocytic and granulocytic myelo proliferation (PMGM) and no or relative reduction of erythroid precursors. Abnormal clustering and increase in atypical giant to medium sized megakaryocytes containing bulbous (cloud-like) hypolobulated nuclei and definitive maturation defects. |
C |
Clinical Stage of PMF or PMGM |
MF |
Staging of myelofibrosis (MF) |
C1 |
Early clinical stages
Normal hemoglobin or slight anemia, grade I: hemoglobin > 12g/dl.
Slight or moderate splenomegaly on ultrasound scan or CT.
Thrombocytosis, platelets in excess of 400, 600 or even 1,000 × 109-1
Normal or increased LAF-score
No leuko-erthroblastose |
MF-0 |
Prefibrotic stage PMF or PMGM: no reticulin fibrosis. |
MF-1 |
Early PMF or PMGM: no reticulin fibrosis. |
C2 |
Intermediate clinical stage
Anemia grade II: hemoglobin > 10g/dl
Definitive leuko-erythroblastic blood picture and/or tear drop erythrocytes increased LDH
Splenomegaly |
MF-1 |
PMF or PMGM slight reticulin fibrosis. |
MF-2 |
Fibrotic PMGM or PMF: marked increase in reticulin and slight moderate collagen fibrosis. |
C3 |
Advanced clinical stage
Anemia grade III: Hemoglobin < 10g/dl
Definitive leuko-erythroblastic blood picture and/or tear drop erythrocytes.
Splenomegaly, Thrombocytopenia, leukocytosis, leukopenia |
MF-3 |
Fibrotic PMGM or PMF: advanced collagen fibrosis with optional osteosclerosis. |
The combination of A1 + B1 establishes PMF or PMGM: -any other criterion C or MF contributes to staging.
Polycythemia vera, a trilinear MPD
A definite diagnosis of PV can be made: plethoric appearance, splenomegaly, definitely elevated erythrocyte count above 6 × 1012/L, elevated platelet count, and elevated hematocrit [1]. The bone marrow is pathognomonic diagnostic showing a panmyelosis (increased trilinear hematopoiesis) and large megakaryocytes (Table 1) [1,2]. The majority of PV patients have a long life span and every attempt should be made to keep the treatment of PV as physiologic as possible by venesection aiming at haematocrit of 0.40 as a satisfactory method resulting in a state of iron deficiency [1-3]. During complete remission of PV by phlebotomy alone red cell count remains elevated above 6 × 1012/L due to microcytosis of red cells, haemoglobin and hematocrit levels remain low for periods of months to years and the PV patient is reasonable asymptomatic indicating that the best index of phlebotomy therapy is the hematocrit value and haemoglobin concentration [2,3]. It is possible to control PV patients by phlebotomy alone for several up to ten to fifteen years and such PV are in as good health as comparable persons of the same age group [3-5].
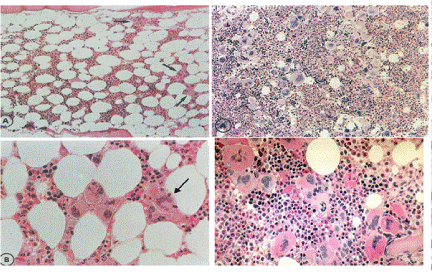
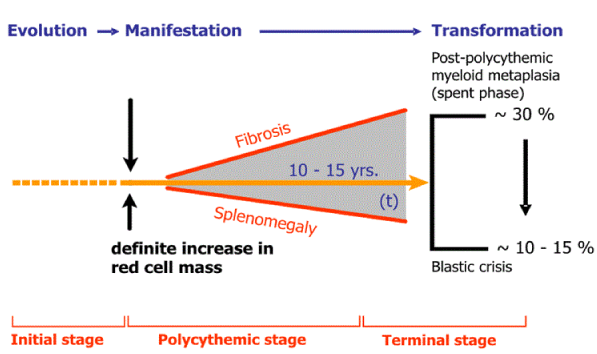
PV as a total marrow disorder in which peripheral blood erythrocytosis, leukocytosis and thrombocytosis are all simultaneously present and the bone marrow is featured by a trilinear myeloproliferaive disease (MPD) of erythrocythemic, thrombocythemic and granulocythemic myeloproliferation (Figure 1-4) [2,3]. PV is complicated by primary myeloid metaplasia of the spleen with increasing degree of splenomegaly, myelofibrosis and the development of anemia in about one third of the cases after long-term follow-up of about 15 to 30 years. The one cause hypothesis cause of Dameshek for PV as a trilinear myeloproliferative disease (MPD) either the presence of excessive bone marrow stimulation by an unknown factor or the lack or diminution of an inhibitory factor has been confirmed by Vainchenker’s discovery of the acquired somatic JAK2V617F mutation as the cause of three phenotypes of MPD essential thrombocythemia (ET), PV, myeloid neoplasia of the spleen (MNS) and secondary myelofibrosis (MF) [2,3].
MPD a benign proliferation of hematopoiesis vs. CML a malignant neoplasia
In 1951 Dameshek speculated on an unifying theory that erythroleukemia, chronic granulocytic leukemia (CML), PV, idiopathic or agnogenic myeloid metaplasia (AMM) of the spleen, and megakaryocytic leukemia (ML) [6,7]. It is possible that these various ‘myeloproliferative disorders” are all somewhat variable manifestations of proliferative activity of the bone marrow cells, or may represent one myeloproliferative activity of bone marrow cells due to a hitherto undiscovered hypothetical stimulus [6]. This concept of MPD is the basis of which the Polycythemia Vera Study Group (PVSG) defined in 1975 the authorative criteria for the diagnosis of PV (Table 1), AMM and primary hemorrhagic thrombocythemia (PHT) and primary myelofibrosis (PMF) [8-10]. PMF or AMM is a clinicopathological entity not preceded by any other PVSG defined MPD PHT or ET, PV, CML or preleukemia with myelodysplastic features. CML is leukemia or neoplasia that destroys normal hematopoiesis whereas ET, PV and AMM form a benign proliferation of trilinear hematopoietic proliferation in the bone marrow (myeloproliferation) and extramedullary hematopoiesis in the spleen [8].
PVSG clinical criteria for PV diagnosis: RCM vs. bone marrow histology
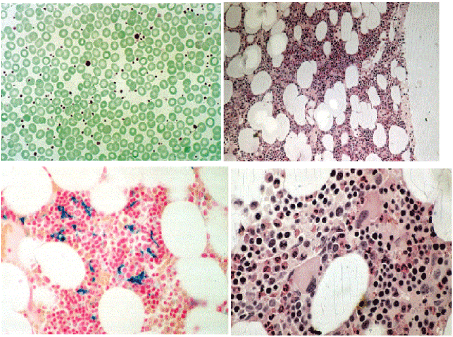
The PVSG defined laboratory findings of 325 PV patients in the PVSG 01 study all PV patients had increased red cell mass by definition and showed an increase in hematocrit > 0.52 in 92%, white cells > 12 × 109/L in 43%, platelets > 400 × 109/L in 63%, leukocyte alkaline phosphatase (LAP) score > 100 in 70% and increased spleen size on palpation (splenomegaly due to myeloid metaplasia) in 70% [10]. Absence of splenomegaly was noted in about 30% of cases and leucocytosis and platelet counts can remain normal in early stage PV with typical PV bone marrow histology [10-41]. Such masked cases of PV with normal platelets, leukocytes and spleen were labeled in 1979 as idiopathic erythrocythemia (IE) by Pearson and Wetherley-Mein [42]. The 1975 PVSG criteria for PV in Table 2 exclude per definition IE (stage 1 PV) [10-41]. IE is featured by increased red cell mass, normal spleen size, normal leukocyte and platelet counts and no clinical or laboratory evidence of primary or secondary erythrocytosis [42]. Leucocyte alkaline phosphatase (LAP) scoring [8,17], in vitro cultures of erythroid progenitors (EEC) [43] and radioimmunoassay of erythropoetin (EPO) do contribute to differentiate PV from all variants erythrocytosis. These assays are useful when there is only isolated elevation of the red cell mass and all the usual causes of secondary polycythemia have been excluded. The characteristic histology findings in bone marrow biopsies of 155 evaluable PV patients with a documented increased RCM in the PVSG 01 study revealed a broad spectrum of bone marrow cellularity from 50% to 60% in 10 cases, from 60% to 80% in 45 cases, and from 80% to 100% in 100 cases (Figure 5) [41]. Silver stained reticulin fiber content was normal (RF-0 and 1 = prefibrotic) in 94 cases, slightly increased (RF-2 = early fibrotic) in 40 cases, and moderately to marked increased (RF-3) in 21 cases. The bone marrow histology diagnoses in the PVSG-01 study [11] could roughly be interpreted as typical for normocellular ET in 10, for PV (hypercellular 60%-80%) in 45, for trilinear PV in 70 and for PV/RF-3 or 4 in 13 PV patients (Table 2, Figure 5).The bone marrow histology data of megakaryopoiesis in PVSG PV and PHT studies were identical in appearance, and the condition PV versus ET could not be distinguished on megakaryocyte histology [39-41]. Increased bone marrow cellularity due to increased erythropoiesis and/or myelopoesis in PVSG defined PV and PHT or ET was identical. The PVSG concluded that the condition PV versus ET could not be distinguished on the basis of bone marrow histopathology. Consequently, the PVSG only used increase red cell mass (RCM) and did not include bone marrow histology as the determinative major inclusion criterion for the diagnosis of PV, and to separate ET from PV [10,39-41]. RCM is insensitive and not specific for the diagnostic differentiation of PV, IE, SE and inapparent PV (Table 1) [17]. In contrast, EEC and bone marrow histology are specific clues for the diagnosis of PV since the early 1970s [1-3,41-43].
From 1988 to 1994, Lamy et al., [44] measured RCM in 103 consecutive PV patients seen in a single center diagnosed as inapparent PV (IPV) in 18 patients (17%) and as PV with increased hemoglobin (Hb) and hematocrit (Ht) defined, respectively, by Hb > 18 g/dL, Ht > 0.52 in males and Hb > 16 g/dL, Ht > 0.47 in females (Table 3) [44]. IPV or masked PV was defined by a normal Hb and Ht value at diagnosis. In the IPV group, the reasons to perform RCM were as follows: splenomegaly associated with increased platelets and/or leucocytes counts (n = 8), portal vein thrombosis (n = 5), increased platelets or leucocytes counts without splenomegaly (n = 3), and isolated splenomegaly (n = 2) [22]. The two groups were balanced in terms of age, sex, leucocyte, serum iron, and platelet level. Hemoglobin, Ht levels, red cell counts, and plasma volumes were significantly different between the two groups (Table 3). Red cell mass was increased in the two groups due to hypervolumemia in PV, but caused by splenomegaly in cases with IPV, whereas the erythrocyte counts were increase in the majority of 85 PV patients, but completely normal in 18 IPV patients except one (Table 3). Consequently IPV with normal or decreased hemoglobin, hematocrit and erythrocytes in Table 3 cannot become candidates for phlebotomy because of absence of hypervolumemic symptoms. Treatment will hydroxyurea carry the great danger of inducing relative anemia and acceleration of myelofibrosis. In the context of splanchnic vein thrombosis (portal or splenic vein thrombosis), and hepatic vein thrombosis (Budd Chiari syndrome), RCM and plasma volume are increased due to splenomegaly as the cause of IPV or masked PV [45].
The 1975 PVSG defined clinical criteria for PV are relatively simple to implement but rather crude thereby overlooking prodromal and masked PV because bone marrow histology as a pathognomonic clue to PV and ET was not considered (Table 1). The PVSG 01 study randomized 431 PV patients for phlebotomy in 134, chlorambucil in 141 and P32 in 156, there was a significant loss of survival of PV patients due to major thrombotic complications during the first 3 years in the phlebotomy arm due to uncontrolled thrombocythemia and aiming at a too high haematocrit just below 0.50 [9,10,49,50]. In retrospect this would not be the case with the recommendation of phlebotomy aiming at a haematocrit of 0.40 according to Dameshek and Pearson et al., [42] on top of aspirin in the United Kingdom and The Netherlands since 1985 [51]. There was a striking increased incidence of overall malignant complications in PV patients with P32 and chorambucil as compared to the phlebotomy-treated PV patients during long-term treatment [49,50]. The overall incidence of leukemia/lymphoma and cancer after 10 to 11 years follow-up was 25% in the phlebotomy arm, 40% in the P32 arm and 67% in the clorambucil arm. The increased incidence of malignancies of bone marrow, lymphoid tissue, skin, and gastrointestinal tract highlights mutagenic effects of chronic myelosuppressive agents in particular when treatment is already started in newly diagnosed early and overt stages of PV. The PVSG 01 trial confirmed the hypothsis of Dameshek [3,50,51] that P32 is leukemogenic when used as the first line myelosuppressive treatment in early and overt stage PV indicating the need to postpone myelopsuppresive therapy in PV as long as possible [52-55]. A large group of low risk PV patients included in the PVSG 01 study were exposed to the leukemogenic agents P32 and chlorambucil. The PVSG 01 investigators recommended around 1990 to replace P32 by hydroxyurea as the first treatment option in PV patients [52-55]. The final analysis of the French PVSG study compared HU (n = 136) vs pipobroman (n = 149) as first-line therapy in 285 newly diagnosed PV patients younger than 65 years (27 patients were older than 65 years) [56]. During follow-up 42 patients (31%) switched from HU to pipobroman because of HU toxicity and 19 (13%) from piopobroman to HU [56]. According to the intention to treat (ITT), the median survival was 17 years for the whole cohort, 20.3 years for the HU arm, and 15.4 for the pipobroman arm (P = 0.008). At 10, 15 and 20 years, the cumulative incidence (probability) of AML/MDS was 6.6%, 16.5% and 24% in the HU arm versus 13%, 34% and 52% in the pipobroman arm. The cumulative incidence (probability) of myelofibrosis (MF) at 10, 15 and 20 years was 15%, 24% and 32% in the HU arm versus 5%, 10% and 21% in the pipobroman arm (P = 0.02) [56]. Results from PV patients who received only one treatment during the entire period (HU n = 94, Pipobroman n = 130) the cumulative incidence of AML/MDS at 10, 15 and 20 years was 7.3%, 10.7% and 16.6% for HU vs 14.6%, 34% and 49.4% for pipobroman [56].
The baseline risk of leukemic transformation in 459 PV and 605 ET patients treated in a single institution retrospective study without cytoreductive therapy or hydroxyurea alone was 3.3% and 7.4%, respectively [57,58]. A primary rigid venesection regimen according to the London PV study group in the late 1970 [59-61] aiming at a hematocrit around to below 0.40 irrespective of gender on top of low dose aspirin introduced by Michiels et al., [51] in 1985 [62-64] will reduce the cumulative incidence of minor circulatory ischemic events and major thrombosis from above 50% to less than 2% per patient/year during long-term follow-up [59-64]. Hydroxyurea should be postponed in early and intermediate stage PV as long as possible by phlebotomy on top of low dose aspirin [17,65]. Current risk stratification in PV and ET should not anymore be based on age above 60 years and history of thrombosis [55], but on real life MPN disease burden using objective parameters including the degree of leucocytosis, splenomegaly, JAK2 allele burden, itching and constitutional symptoms [65]. Hydroxyurea (HU) and JAK2 inhibitors are indicated according to not yet clearly defined recommendations in hyperproliferative PV with increased to high MPN disease burden althought targeted recommendations on the use of JAK2 inhibitors will be further clarified and defined soon [66].
The 1975 PVSG criteria for ET
Primary hemorrhagic thrombocythemia (PHT) has already been defined in 1960 by Gunz as clinical syndrome of recurrent spontaneous hemorrhages often preceded by thromboses, extremely high platelet count in excess of 1000 × 109/L, frequently splenomegaly, and hypochromic anemia with a tendency towards polycythemia between hemorrhages [37]. Mucocutaneous bleeds from nose gums and gastrointestinal tract were most frequent followed by bruises and bleedings after trauma or surgery [38]. Accordingly, the PVSG used from 1975 to 1986 a minimum platelet count of 1000 × 109/L for the diagnosis for PHT without features of PV or AMM [8,38-40]. The PVSG inclusion and exclusion criteria in 1975 for the diagnosis of PHT or essential thrombocythemia (ET) were very crude [8]: (1) A platelet count in excess of 1000 × 109/L and a bone marrow smear which shows marked megakaryocytic hyperplasia and abundant platelet clumps; (2) Absence of polycythemia vera as defined by the PVSG (normal red cell mass (RCM) [10]; (3) Absence of the Philadelphia chromosome to exclude CML [16]; and (4) Absence of significant reticulin fibrosis (myelofibrosis) with dry tap on bone marrow aspiration, and no signs of preleukemia erythroleukemia or trilinear myelodysplastic syndrome [8,39,40].
PVSG defined PHT8 labeled as ET [39,40] is featured by platelet counts between 1000 and 3000 × 109/L, splenomegaly in about 80%, autoinfarction of the spleen in 20%, and iron deficient microcytic anemia in 60% [39,40]. In 40% of PHT patients gastrointestinal roentgenograms suggest duodenal ulcer caused by small infarcts in the duodenal mucosa resulting from the high platelet count [40]. In the first prospective evaluation of PVSG defined PHT, 37 evaluable ET patients with platelet counts between 1000 to 2650 × 109/L suffered from thrombohemorrhagic events at presentation including mild bleedings in 5 epistaxis in 5, ecchymoses in 2, pelvic, buccal, fundal or urinary tract hemorrhage in 6, melena with a fall in hemoglobin of 7 gm/dL in 1 and massive postoperative bleeding in 1 case [39,40]. Eleven ET patients experienced acroparesthesias (numbness), including burning sensations, usually in hand or feet (suggestive for erythromelalgia), 9 had dizziness, light-headedness or syncope, 7 had visual disturbances such as scotomas and transient dimming or blurred vision. Catastrophic complications (severe hemorrhages, myocardial infarction, stroke) in 6 (16%) were observed. In this study of 37 untreated PHT or ET patients, bone marrow cellularity was normal in 11%, increased between 50% to 90% in 78% and greater than 90% in 11% [39,40]. Two-thirds of diagnostic bone marrow biopsies showed marked megakaryocyte hyperplasia with atypical large megakaryocytes. Reticulin content was essentially normal in 90% indicating prefibrotic MPD. MPD features in ET and PV were quiete similar: leukocytosis was common in PHT (ET) and PV, LAP scores over 100 were seen in 42% of PHT, and in 70% of PV patients; pruritis was observed in 14% in PHT and 43% in PV patients; the spleen was palpable in 38% of PHT and 70% of PV patients, and when enlarged in PHT the spleen was palpable 2 to 4 cm below the costal margin [8]. Since 1975 we discovered a causal relation between erythromelalgic microvasclur disturbances and thrombocythemia in early early stage MPD disease at platelet counts above 400 × 109/L in symptomatic ET and PV patients with persistent increased platelet count in excess of 400 × 109/L and recognized that the increase of clustered large pleomorphic megakaryocytes in bone marrow biopsies was a pathognomonic clue to myeloproliferative thrombocythemia in ET and PV (Table 4).
The 1980 Rotterdam clinical and pathological (RCP) criteria for ET and PV
As the 1975 PVSG criteria for ET (PTH) are crude, we could recognize since 1975 the existence of erythromelalgic thrombotic thrombocythemia (ETT) in early stage MPD with a persistent increase of platelet count in excess of 400 × 109/L [17,81,82]. At that time we followed since 1975 the definition of PV according to Dameshek as a trilinear MPD [2,3] of which the bone marrow features were described as erythrocytic megakaryocytic granulocytic myeloproliferation (EMGM) by Kurnick et al., [83] in 1972 and Ellis et al., [41] in 1975 (Figure 6). The trephine biopsy in our ET cases showed a proliferation of large mature megakaryocytes in a normal cellular bone marrow with normal erythropoiesis and granulopoiesis (Table 4, Figure 7). In PV increased erythropoiesis was most prominent [76] together with variable degrees of increased platelets (> 400 × 109/L) [10], erythrocytes (> 6 × 1012/L) [2,3] and granulocytes in the peripheral blood in the absence of the Ph-chromosome (Table 4) and could document distinct bone marrow features as the pathognomonicclue to very early stage of ET (Figure 7). The 1980 RCP criteria of ET and PV were determined by careful prospective documentation of peripheral blood and bone marrow smears and bone marrow biopsy material (Table 4). Platelets in excess of 400 × 109/L, and an increase of clustered enlarged megakaryocytes in a bone marrow biopsy material was found to be pathognomonic diagnostic for ET and excluded reactive thrombocytosis. The combination of bone marrow histology and erythrocyte count above 6 × 1012/L according to Dameshek [1-3] appeared to be specific clues to the diagnosis of PV (Table 3) clearly different from all variant of primary and secondary erythrocytosis as documented by Kurnick et al., [83] in 1972 and by Vykoupil, Thiele, Stangel, Krompotic and Georgii in 1980 [67]. The 1980 RCP modifications of the 1975 PVSG criteria for PV include 4 main changes (Table 4). First, the major criterion O2-saturation of > 92% is replaced by absence of primary or secondary erythrocytosis by clinical and laboratory tests. Second; splenomegaly is replaced by bone marrow histology as a major criterion (A3). Third, the 1980 RCP diagnostic set used splenomegaly as a minor criterion (Table 4). Fourth, we skipped raised B12 (> 900 ng/L) or raised B12 binding capacity (> 2200 ng/L) as completely irrelevant for the diagnosis of early and overt stage PV (Table 4).
Between 1975 and 1980, we prospectively evaluated the RCP criteria in 30 consecutive early prefibrotic stage patients, who presented with erythromelalgic thrombotic thrombocythemia (ETT), 14 ET and 16 PV patients [84]. The mean age of 30 ETT patients (ET and PV) was 56.7 (range 33-96) years. Eleven of 14 ET patients had platelet counts below 1000 × 109/L, in whom the diagnosis of ET would have been overlooked by the crude PVSG criteria at that time between 1975-1985. Spleen size on scan was slightly increased in 5 of 14 ET and in 13 of 16 PV patients. Leukocyte count counts was increased (> 10 × 109/L) in 5 out of 14 patients with ET and in 14 of 16 PV patients. LAP score was increased (> 100) in 12 out of 14 ET and in all PV patients (Figure 8). All PV patients with erythrocytes above 6 × 1012/L had increased red cell mass (manuscript in preparation). Increased erythrocyte counts above 6 × 1012/L and increase of large pleomorphic megakaryocytes in bone marrow smear (Dameshek) [2,3] and biopsy is diagnostic for PV [17]. Erythrocyte count at a cutoff level of 6 × 10/12L differentiates ET from PV on top of a typical MPD bone marrow histology obviating the need to measure RCM (Table 4). Increased erythrocytes above 6 × 1012/L persists in PV in a comple hematological remission by repeated venesection [2,3,77,78].
The presence of clustered large pleomorph megakaryocytes in bone marrow smears and biopsies is diagnostic clue for ET and PV (Table 4, Figures 6 and 7). An ET bone marrow picture with increase of clustered pleomorphic megakaryocytes and no increase of cellularity (Figure 7) was seen in 7 of 14 ET and only in 1 of 16 PV patients [83,84]. A moderate increase of cellularity (60%-80%) in the bone marrow due to increased erythropoiesis (= decreased M/E ratio) consistent with early stage PV was seen in 3 ET and 4 PV patients. A typical PV hypercellular (80%-100%) bone marrow due to megakaryo/erythro/granulocytopoiesis (Figure 6) was seen in one ET patients and in the majority of PV [83,84]. These results indicate that bone marrow histopathology on its own is characteristic for MPN but not fully reliable to differentiate between ET and PV [77,78]. The peripheral blood findings in 30 ET (primary thrombocythemia; PT) and 20 PV) seen between 1975 and 1985 are shown in Figure 7 [84]. Hemoglobin levels are normal in ET and elevated in PV, leukocytes were normal or elevated in ET and PV [84]. Out of 30 ET patients 24 had an increased and 6 had a normal LAP scores (Figure 8). Platelet counts were were between 400 × 109/L and 1000 × 109/L in the majority of the 30 ET patients. Serum uric acid levels were normal in ET and frequently elevated in PV. Increased LAP score in the absence of infection and normal erythrocyte sedimentation rate (ESR) or C-reactive protein (CRP) is indicative for MPN PV or ET. LAP score was much more elevated in all 20 PV than in ET [84]. As leucocyte alkaline phosphatase (LAP) scoring is becoming increasingly rare in common practice, it can easily be replaced by CD11b neutrophil expression [85]. In the setting of SVT patients PV patients showed higher CD11b values of 190 (CI: 151-238) in PV versus 111 (CI: 81-153) in non-PV patients with SVT [85]. In routine practice CD11b expression in the MPNs PV, ET of various molecular etiology vs all variants of erythrocytosis should be assessed aganist CD11b expression in controls who have normal ESR and CRP in order to kick out false positive due to underlying infectious or systemic diseases.
The 1990 Hannover Bone Marrow Classification of the MPDs
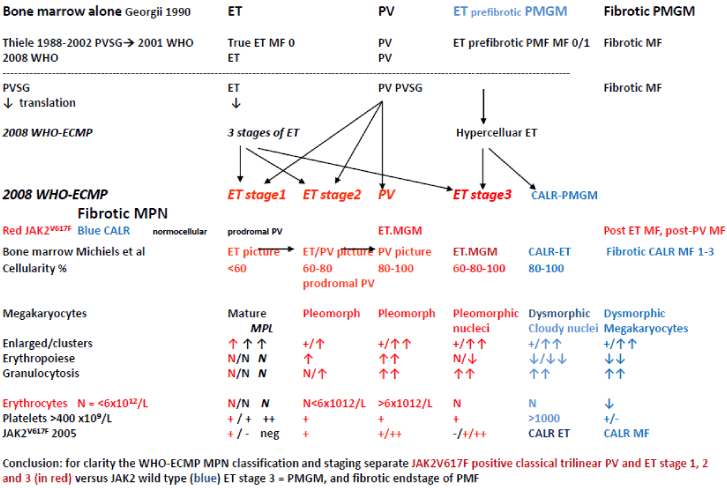
The PVSG distinguished four distinct categories of myeloproliferative disorders (CML, ET, PV and AMM or PMF) and a fifth category of unclassifiable MPD. Since 1976 the German pathologists Burkhardt and Georgii had clearly defined the pathological features of ET, PV and CMGM [67-80] on the basis of which we could developed the European Clinical and Pathological (ECP, Table 5AB,6 and 7) criteria for ET and PV [81,82]. The concept of this traditional classification by the PVSG and the WHO in 1979 has been revised by Georgii in his Hannover Bone Marrow classification of the MPDs as follows: primary diseases are CML, PV, thrombocythemia, chronic megakaryocytic granulocytic myeloproliferation (CMGM) and unclassifiable MPD [35-37]. Reticulin and collagen fibrosis is a reactive feature consecutive to myeloproliferation [37]. The reliable distinction within the three Ph-negative MPDs and its variations appears to be problematic caused by an overlapping cytomorphology of megakaryocyte and within the three MPDs thrombocythemia, PV and CMGM. According to Georgii & Michiels an MPD classification system should be focused cytomorphology of megakaryocytes from bone marrow smears and histopathology from bone marrow biopsies [30-32,35,36]. Megakaryocyte morphology and bone marrow histology had become a hallmark of distinction for the diagnostic differentiation of thrombocythemia (ET), PV versus CMGM in the Hannover Institute of Pathology since 1980 (Figure 5) [67,68]. For the understanding of MPD classification bone marrow histology should distinguish between primary prefibrotic and advanced diseases [35,36]. Primary prefibrotic MPDs according to the Hannover Bone Marrow Classification in 1990 include PV, normocellular thrombocythemia and hypercellular thrombocythemia associated with CMGM without any feature of PVSG defined PV [35,36]. Results from Adamson et al., [69], Fialkov et al., [70,71] and many others revealed the reactive nature of reticulin and collagen fibrosis within the MPDs [69-74]. Consequently, the term of chronic idiopathic myelofibrosis (CIMF) or primary myelofibrosis (PMF) cannot be considered as correct in the Hannover Bone Classification of the MPDs since it has to be considered as a consequence of an underlying myeloproliferative disease [35-37,73,74]. In the 1990 Hannover bone marrow classification, Georgii omitted the terms AMM, CIMF and PMF and introduced the entity of CMGM as the third distinct entity without features of PV and ET [57,58]. The diagnosis of prefibrotic CMGM is mainly based on the presence of large immature megakaryocytes with immature cytoplasm and cloud-like nuclei not seen in ET and PV (Figure 5) [35,36]. The term CMGM has illogically been changed into CIMF under the influence of Thiele and Vardiman in the 2001 WHO classification [75] and as primary myelofibrosis (PMF) in the 2008 WHO classification [76]. With the advent JAK2 and MPL mutations as driver causes of clonal ET and PV Michiels et al., [77,78] recognized CMGM as the third distinct MPD entity and replaced the term CMGM by JAK2/MPL wild type primary megakaryocytic granulocytic myeloproliferation (PMGM) in the 2002-2005 ECP (Table 5AB, 6 and 7), and the 2007 ECMP and 2015 WHO-CMP classifications (Tables 8-11).
The 2000-2005 ECP of thrombocthemia in various MPDs
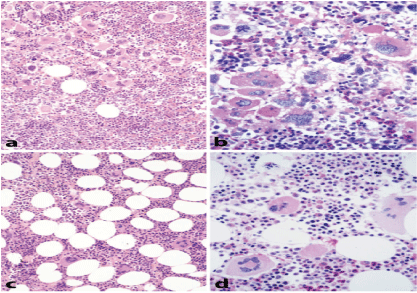
In 2002 Michiels and Thiele defined “true” ET and differentiated “true” ET from hypercellular ET associated with prefibrotic CIMF/PMGM (Table 5AB, Figure 9) [82]. In “true” ET megakaryocytes display large to giant megakaryocytes showing hyperlobulated staghorn-like nuclei in a normocellular bone marrow, (Table 6, Figure 9) [78,80]. PV was typically featured by large pleomorphic megekaryocytes with hyperploid nuclei in a hypercellular bone marrow due to increased erythropoiesis or increased erythrocytic-megakaryocytic-granulocytic myeloproliferation. Interestingly the megakaryocytes in “true” ET were larger than in PV (Table 5A, Figure 9) [1]. Hypercellular ET associated with prefibrotic CIMF (“false” ET = CMGM = PMGM, (Table 5B, Figures 5 and 9) is dominated by an increase of clustered atypical dysmorphic megakaryocytes due to increases of cellular and nuclear size and bulky nuclei with clumsy lobuli and irregular roundish shaped form (so-called cloud-like nuclei, Figures 5 and 9), which are never described in ET and PV2. Normocellular “true” ET according to the 2002 ECP criteria is featured by normal LAP scores (normal CD11b neutrophil expression), higher platelet counts and large to giant megakaryocytes with multilobulated stag-horn like nuclei in a completely normocellular bone marrow (Table 5A) [90,91]. In “true” ET the values for hemoglobin, erythrocytes, and LAP scores (CD11b neutrophil expression) were completely normsal (Table 5A). In contrast, normocellular ET and prodromal PV with increased LAP score (Figure 8) show the presence of pleomorphic megakaryocytes (Figure 6) low serum EPO levels, slight splenomegaly, and spontaneous EEC [17,84]. This point to the existance of at least two phenotypes of normocelluar ET: ET with features of early PV (prodromal PV) versus “true” ET without features of PV. These differences in RCP defined ET (Table 1) criteria and ECP-defined “true” ET and false ET (Table 5A) are related to a selection bias of patients. In 1988, Thiele as a pathologist selected 25 cases with 1975 PVSG defined normocellular ET (minimum platelet count of 1000 × 109/L) who had pronounced thrombocythemia and normocellular bone marrow without PV features and this was associated with normal LAP score (CD11b neutrophil expression) [87]. Focussing on aspirin-sensitive erythromelalgic inflammatory and ischemic complications as pathognomonic presenting symptoms of early ET or PV patients, the Rotterdam MPD Working group studied a selected and biased group of early stage myeloproliferative ET and PV at platelet counts above 400 × 109/L [84]. Only symptomatic ET and PV patients were included in our 1975-1985 studies because of erythromelalgic ischemic digital circulation disturbances [44].
The 2006-2015 WHO-ECMP Criteria for MPD/MPN
Myelofibrosis is not specific for a disease and can be observed in patient with hairy cell leukemia, Ph-positive CML and in the Ph-negative MPDs , CMGM, PV, ET and .many other conditions as well [35-37,92,93]. An increase of reticulin in the silver impregnation stain of bone marrow biopsies can be graded as fine or coarse fibers and is a secondary event relevant for prognosis assessment (Table 2) [94]. The use of CIMF and PMF in the 2001 and 2008 WHO classifications [75,76,94-96] is misleading and scientifically not recommended [94]. At that time we had to use the term CIMF in our publications on the 2006/2007 WHO-ECMP classification [94-96] for reasons of compliance with peer reviewers, but we convincely eliminated the terms CIMF and PMF by primary megakaryocytic granulocytic myeloproliferation (PMGM) in the publication on the 2007 ECMP and 2015 WHO-CMP classifications (Tables 8-11).With the advent of the JAK2 mutation for the trilinear MPNs we separated the JAK2V617F positive ET, PV (or EMGM) from the JAK2-negative normocellular ET and hypercellular ET associated with PMGM (Tables 8-11) [77,78,94-96]. Myelofibrosis (MF) itself is the reflection of MPN disease progression but not a disease entity on its own because reticulin and collagen fibrosis are produced by polyclonal fibroblasts in response to cytokines released from the clonal granulocytic and megakaryocytic proliferative cells in both PV and ET of various molecular etiology [69-74]. The presence of reticuline fibrosis is well documented in all variants of ET, PV, PMGM, CML and in many other conditions. An increase of reticulin fibrosis is rare in WHO normocellular ET will occur in about one third of PV and will occur in the majority of patients with PMGM during long-term follow-up [35,36,89].
The WHO bone marrow features and the variable phenotypes of thrombocythemias according to the 2000-2005 ECP and the 2006/2007 WHO-ECMP criteria for ET do in fact distinguish within the JAK2V617F mutated MPN normocellular (WHO-ET, < 60%) and hypercellular ET (prodromal PV, 60%-90%) due to increased erythropoiesis (prodromal PV), from JAK2 wild type normocellular ‘true’ ET (Tables 5A) and from MPL515-mutated normocellular ET (Table 10). We separated JAK2 mutated MPN from JAK2 wild type hypercellular ET associated with a granulocytic myeloproliferation (PMGM, Table 11, Figure 10) [94-96] JAK2V617F mutated normocellular ET and prodromal PV patients had increased LAP score similar as in PV (Figure 8). Bone marrow histopathology on its own was not reliable to differentiate between JAK2V617F positive ET, prodromal PV and classical PV. The 200-2005 ECP criteria had defined “true” ET with normal LAP score and giant megakaryocytes with staghorn-like nuclei in a normocellular bone marrow (Table 5) proved to be consistent with MPL515 mutated normocellular thrombocythemia (Table 10). Prefirotic CIMF = PMF = PMGM (Table 11) belonged since 2005 to the JAK2/MPL wild type MPD/MPN (Tables 5B, Table 11, Figure 10) [64], and proved to become thecalreticulin (CALR) mutated Thrombocythemia and MF in the 2015 WHO-CMP claasification (Table 11).
According to Michiels and Thiele, WHO bone marrow biopsy histopathology evaluations have a sensitivity and specificity near to 100% to differentiate all variants of ET in various MPD/MPNs from thrombocythemia in CML, from myelodysplastic syndromes and from reactive thromcytoses [91,94-96]. The distinction of prefibrotic thrombocythemias in the MPNs of various molecular etiology at the bone marrow level is a topic of research for the Dutch and Belgian hematopathologists [77,78,94-96] and not discussed in this insight review. The ECP, ECMP and WHO bone marrow features separate myeloroliferative PV from all variants of primary congenital erythrocytoses and secondary erythrocytoses [77,78]. Bone marrow histology separates idiopathic erythrocytosis with increased RCM from early erythrocythemic stage 1 PV, and do detect “masked” ET, “masked” PV overlooked by the PVSG and WHO criteria with a sensitivity and specificity of 100% if the trephine biopsy is of good quality [77,78]. A bone marrow biopsy is mandatory for grading cellularity in prefibrotic stages and for grading reticulin and collagen fibrosis (Tables 1 and 7) in hypercellular MPN advanced PV and ET (post-ET myelofibrosis, post-PV myelofibrosis).
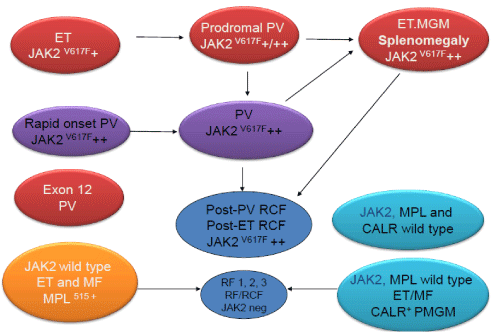
The formulation of the ECMP criteria in 2006/2007 by Michiels et al., [94-96] (Tables 8-11, Figure 10) for ET prodromal PV and PV preceded the publication of the revised WHO classification of the Myeloproliferative Neoplasms (MPN) in 2007 and 2008 [96-98]. Thiele et al., validated the 2008 WHO classification of MPN in JAK2V617F positive and JAK2 wild type MPN from large series of PVSG defined MPD patients diagnosed in the past and persisted to use the terms ‘’true” ET versus “false” ET or PMF [99-102]. The 2008 WHO investigators were persistently confronted with unsolved problems and pitfalls regarding in ET of various MPNs and did not recognize the distinct differences in bone marrow histology between JAK2 mutated ET and PV and JAK2 wild type ET without features of PV. The 2008 WHO recognized the differences in megakaryocyte morphology in CML, MDS as compared to PV, but did not pick up the potential importance of significant differences of megakaryocyte morphology related to JAK2 and MPL mutated versus JAK2/MPL wild type ET and MF [77,78]. The 2007-2015 WHO and European clinical, molecular and pathological (EuroCMP) classification of the prefibrotic MPNs distuinguishes JAK2V617F mutated trilinear ET, prodromal PV, masked PV and classical PV (Figure 11). Based on distinct megakaryocyte morphology features JAK2 wild type “true” ET (Table 10) carrying the MPL mutation clearly differs from JAK2/MPL wild type hypercelluar ET associated with CMGM/PMGM (Table 11, Figure 10). JAK2 wild type MPL515 mutated ET is the second distinct thrombocythemia featured by clustered giant megakaryocytes with hyperlobulated stag-horn-like nuclei, in a normocellular bone marrow consistent with the diagnosis of “true” ET. JAK2/MPL wild type hypercellular ET is the third distinct thrombocythemia characterized by clustered larged immature dysmorphic megakaryocytes and bulky (bulbous) hyperchromatic nuclei consistent with CMGM/PMGM.
The 2015 WHO-CMP criteria define three phenotypes of JAK2V617F mutated Myeloproliferative Neoplasms (MPNs) essential thrombocythemia (ET), prodromal polycythemiavera (PV), prodromal PV, classical PV or erythrocytic, megakaryocytic granulocytic myeloproliferation (EMG) and hypercellular ET associated with EMG (masked PV) versus the JAK2 exon 12 mutated idiopathic eryhrocythemia (IE) and PV (Figure 12) [74,75]. MPL515 mutated JAK2 wild type ET and MF is a distinct thrombocythemia without features of PV in blood and bone marrow (Figure 12). Calreticulin mutated Thrombocythemia and MF is the third thrombocythemia entity with characteristic features of primary megakaryocytic granulocytic myeloproliferation (PMGM) in the bone marrow, which are not seen in JAK2 and MPL mutated MPNs (Figure 12) [103,104]. MPN disease burden in JAK2, MPL and CALR mutated MPN is best reflected by the degree of anemia, splenomegaly, mutation allele burden, bone marrow cellularity and myelofibrosis (Figure 12) [103,104].
- Dameshek W, Henstell HH (1940) The diagnosis of polycythemia. Ann Intern Med 13: 1360-1387.
- Dameshek W (1950) Physiopathology and course of polycythemia vera as related to therapy. J Am Med Assoc 142-790-797.
- Michiels JJ (2013) Physiopathology, etiologic factors, diagnosis and course of polycythemia vera as related to therapy according to William Dameshek 1940-1950. Turkish J Hematol 30: 102-110. [Crossref]
- Dameshek W (1946) The treatment of Polycythemia. Blood 1: 256.
- Erf LA (1946) Primary polycythemia: remissions induced by therapy with radiophosphorus. Blood 1: 202-208. [Crossref]
- Dameshek W (1951) Some speculations on the myeloproliferative syndromes. Blood 6: 372-375. [Crossref]
- Tefferi A (2008) The history of myeloproliferative disorders: before and after Dameshek. Leukemia 22: 3-13. [Crossref]
- Laszlo J (1975) Myeloproliferative disorders (MPD): myelofibrosis, myelosclerosis, extramedullary hematopoiesis, undifferentiated MPD and hemorrhagic thrombocythemia. Semin Hematol 12: 409-432. [Crossref]
- Wasserman LR (1971) The management of polycythemia vera. Br J Haematol 21: 371-376.
- Berlin NI (1975) Diagnosis and classification of the polycythemias. Sem Hematol 12: 339-351. [Crossref]
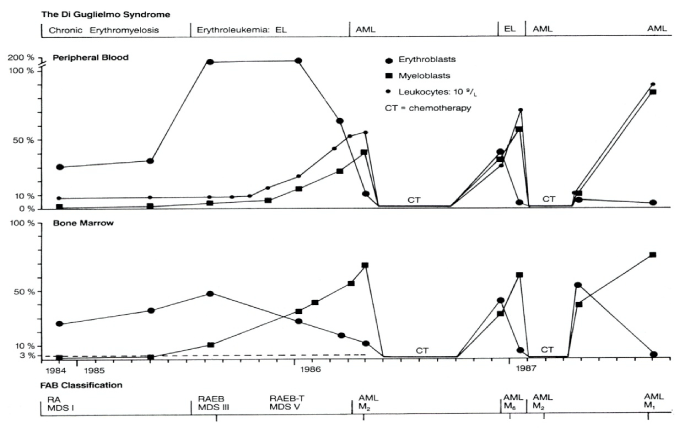
- Dameshek W, Baldini M (1958) The Di Guglielmo syndrome. Blood 13: 192-195.
- Dameshek W (1969) The Di Guglielmo syndrome revisted. Blood 34: 567-572. [Crossref]
- Michiels JJ (1993) Erythroleukemia and myelodysplastic syndromes. An historical appraisal and a personal view. Leukemia & Lymphoma 9: 27-34. [Crossref]
- Greenberg PL, Attar E, Bennett JM, Bloomfield CD, De Castro MD, et al. (2013) Myelodysplastic Syndromes. Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 11: 838–874. [Crossref]
- Nowell PC, Hungerford DA (1960) Chromosome studies on normal and leukemic leukocytes. J Natl Cancer Inst 25: 85-109. [Crossref]
- Gilbert HS (1973) The spectrum of myeloproliferative disorders. Med Clin North Amer 57: 355-393.
- Michiels JJ (1996) The myeloproliferative disorders. An historical appraisal and personal experiences. Leukemia and Lymphoma 22: 1-14. [Crossref]
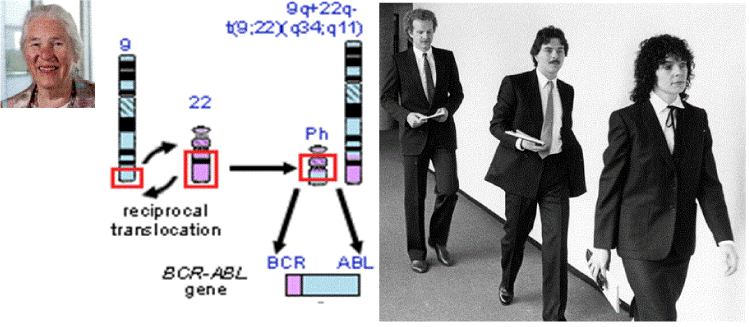
- Rowley J (1973) A new consistent chromosomal abnormality in chronic myelogenous leukemia identified by quinacrine fluorescence Giemsa staining. Nature 243: 290-291. [Crossref]
- Rowley JD (1980) Ph1-positive leukaemia, including chronic myelogenous leukaemia. Clin Haematol 9: 55-86. [Crossref]
- Rowley JD (1984) Biological implication of consistent chromosome rearrangement in leukemia and lymphoms. Caancer Res 44: 3159-3168. [Crossref]
- Heisterkamp N, Groffen J, Stephenson JR, Spurr NK, Goodfellow PN, et al. (1982) Chromosomal localization of human cellular homologues of two viral oncogenes. Nature 299: 747-749.
- De Klein A, Van Kessel AG, Grosveld GG (1982) A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukemia. Nature 300: 765-767. [Crossref]
- Bartram CR, de Klein A, Hagemeijer A (1983) Translocation of the human c-abl oncogene occurs in variant Ph-positive but not Ph-negative chronic myeloid leukemia. Nature 306: 277-280.
- Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, et al. (1984) Philadelphia chromosomal breakpoits are clustered within a limited region, bcr on chromosome 22. Cell 35: 93-99. [Crossref]
- Lugo TG, Pendergast AM, Muller AJ, Witte ON (1990) Tyrosine kinase activity and transformation potency of bcr/abl oncogene products. Science 247: 1079-1082. [Crossref]
- Kelliber MA, McLaughin J, Witte ON, Rosenberg N (1990) Induction of a chronic myelogenous-like syndrome in mice with v-abl and BCR/ABL. Proc Nat Sci USA 87: 6649-6653. [Crossref]
- Daley GQ, Van Etten RA, Baltimore D (1990) Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 247: 824-830. [Crossref]
- Shephard PCA, Ganesan TS, Galton DAG (1987) Haematological classification of the chronic myeloid leukemias. Bailliere's Clin Haematol 1: 887-906. [Crossref]
- Baccarani M, Deiniger MW, Rosti G, Hochhaus Soverini H, Apperly JF, et al. (2013) European LeukemiaNet recommendations for the management of chronic myeloid leukemia. Blood 122: 872-884. [Crossref]
- Michiels JJ, Prins ME, Hagemeijer A, Brederoo P (1987) Philadelphia chromosome-positive thrombocythemia and megakaryoblast leukemia. Am J Clin Pathol 88: 645-652. [Crossref]
- Michiels JJ, Ten Kate FJW (1992) Erythromelalgia in thrombocythemia of various myeloproliferative disorders. Am J Hematol 39: 131-136. [Crossref]
- Michiels JJ, Berneman ZW, Schroyens W, Kutti J, Swolin B, et al. (2004) Philadelphia (Ph) chromosome positive thrombocythemia without features of chronic myeloid leukemia in peripheral blood: natural history and diagnostic differentiation from Ph-negative essential thrombocythemia. Ann Hematol 83: 504-512. [Crossref]
- Thiele J, Schneider G, Hoeppner B, Wienhold S, Zankovich R, et al. (1988) Histomorphometry of bone marrow biopsies in chronic myeloproliferative disorders with associated thrombocytosis – features of significance for the diagnosis of primary (essential) thrombocythemia. Virchows Arch A Pathol Anat Histopathol 413: 407-417. [Crossref]
- Thiele J, Zankovich R, Steinberg T, Kremer B, Fischer R, et al. (1981) Primary (essential)thrombocythemia versus hyperplastic stages of agnogenic myeloid metaplasia (AMM) with thrombocytosis: a critical evaluation of clinical and histomorphological data. ActaHematol 81: 192-202
- [Crossref]
- Georgii A, Vykoupil KF, Buhr T, Choritz H, Döhler U, et al. (1990) Chronic myeloproliferative disorders in bone marrow biopsies. Path Res Pract 186: 3-27. [Crossref]
- Georgii A, Buhr T, Buesche G, Kreft A, Choritz H (1996) Classification and staging of Ph-negative myeloproliferative disorders by histopathology from bone marrow biopsies. Leukemia Lymphoma 22:15-29. [Crossref]
- Michiels JJ, Ten Kate F, Lam KH, Schroyens W, Berneman Z, et al. (2014) The European Clinical Molecular and Pathological (ECMP) criteria and the 2007/2008 revisions of the World Health Organization (WHO) fort he diagnosis, classification and staging of prefibrotic myeloprliferative neolasms carrying the JAK2V617F mutation. Turk J Hematol 31: 136-142. [Crossref]
- Gunz FW (1960) Hemorrhagic thrombocythemia. A critical review. Blood 15: 706-723. [Crossref]
- Iland H, Laszlo J, Peterson P, Murphy S, Briere J, et al. (1983) Essential thrombocythemia: clinical and laboratory characteristics at presentation. Trans Assoc Am Phys XCVI: 165-174.
- Murphy S, Iland H, Rosenthal D, Lazslo J (1986) Essential thrombocythemia: an interim report from the Polycythemia Vera Study Group. Semin Hematol 23: 117-182. [Crossref]
- Ellis JT, Silver RT, Coleman M, Geller SA (1975) The bone marrow in polycythemia vera. Semin Hematol 12: 433-444. [Crossref]
- Pearson TC, Wetherley-Mein G (1979) The course and complications of idiopathic erythrocytoses. Clin Lab Haematol 1: 189-196. [Crossref]
- Pchral JF, Axelrad AE (1974) Bone marrow responses in polycythemia vera. N Eng J Med 290: 1382. [Crossref]
- Lamy T, Devillers A, Bernard M, Moison A, Grulois I, et al. (1997) Inapparent polycythemia vera: an unrecognized diagnosis. Am J Med 102: 14-20. [Crossref]
- De Stefano V, Teofili L, Leone G, Michiels JJ (1997) Spontaneous erythroid coloney formation as the clue to an underlying myeloproliferative disorder in patients wit Budd-Chiari syndrome or portal vein thrombosis. Sem Thromb Hemostas 23: 411-418. [Crossref]
- Briere JB (2006) Budd-Chiari syndrome and portal vein thrombosis with myeloproliferative disorders: diagnosis and management. Sem Thromb Hemostas 32: 208-218. [Crossref]
- Kiladjian JJ, Cervantes F, Leebeek FWG (2008) The impact of JAK2 and MPL mutations on diagnosis and prognosis of splanchnic vein thrombosis: a report on 241 cases. Blood 111: 4922-4929. [Crossref]
- Smalberg JH, Arends L, Valla DC, Kiladjian JJ, Janssen HLA, et al. (2012) Myeloproliferative neoplasms in Budd-Chiari syndrome andportal vein thrombosis: a meta-analysis. Blood 120: 4921
[Crossref]
- Wasserman LR, Berk PD, Berlin NI (1995) Polycythemia vera and the myeloproliferative disorders. WB Saunders Philadelphia.
- Berk PD, Goldberg JD, Silverstein MN (1981) Increased incidence of acute leukemia in polycythemia vera associated with chlorambucil therapy. N Eng J Med 304: 441-447. [Crossref]
- Michiels JJ, Abels J, Steketee J, vanVliet HHDM, Vuzevski VD (1985) Erythromelalgia caused by platelet-mediated arteriolar inflammation and thrombosis in thrombocythemia. Ann Intern Med 102: 466-471. [Crossref]
- Berk PD, Goldberg JD, Donovan PD, Fruchtman SM, Berlin NI, et al. (1986) Therapeutic recommendations in polycythemia vera based on the PVSG protocols. Sem Hematol 23: 132-143. [Crossref]
- Donovan PB, Kaplan ME, Goldberg JD, Tatarsky I, Najean Y, et al. (1984) Treatment of polycythemia vera with hydroxyurea. Am J Hematol 17: 329-34. [Crossref]
- Fruchtman SM, Mack K, Kaplan ME, Peterson P, Berk PD, et al. (1997) From efficacy to safety: a Polycythemia Vera Study group report on hydroxyurea in patients with polycythemia vera. Semin Hematol 34: 17-23. [Crossref]
- Barbui T, Barosi G, Birgegard G (2011) Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 29: 761-770. [Crossref]
- Kiladjian JJ, Chrevet S, Dosquet C, Chomienne C, Rain JD (2011) Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol 29: 3907-3913. [Crossref]
- Gangat N, Strand J, Li CY (2007) Leukocytosis in polycythemia vera predicts both inferior survival and leukemic transformation. Br J Haematol 138: 354-358. [Crossref]
- Gangat N, Wolanski AP, McClure RF (2007) Risk stratification for survival and leukemic transformation in essential thrombocythemia: a single institutional study of 605 patients. Leukemia 21: 270-276. [Crossref]
- Thomas DJ, Marshall J, Russel RWR, Wetherlet-Mein G, du Boulay GH, et al. (1977) Cerebral blood-flow in polycythaemia. Lancet II: 161-163. [Crossref]
- Pearson TC, Grimes AJ, Sender NGP, Wetherley-Mein G (1978) Vascular occlusive episodes and venous haematocrit in primary proliferative polycythemia. Lancet II: 1219-1222. [Crossref]
- Messinezy M, Pearson TC, Prochchazka A, Etherley-Mein G (1985) Treatment of primary proliferative polycythaemia by venesection and lowdosebusulphan: retrospective study from one centre. Brit J Haematol 61: 657-666. [Crossref]
- Landolfi R, Marchio2021 Copyright OAT. All rights reservG, et al. (2004) Efficacy and safety of low-dose aspirin in polycythemia vera: results of the ECLAP trial. New Eng J Med 350: 114-124. [Crossref]
- Landolfi R, Di Gennarro L, Falanga A (2008) Thrombosis in myeloproliferative disorders: pathogenetic facts and speculation. Leukemia 22: 2020-2028. [Crossref]
- Van Genderen PJJ, Mulder PGH, Waleboer M, van de Moesdijk D, Michiels JJ (1997) Prevention and treatment of thrombotic complications in essential thrombocythemia: efficacy and safety of aspirin. Brit J Haematol 97: 179-184. [Crossref]
- Michiels JJ (2014) Erythromelalgic microvascular disturbances, major thrombosis and hemorrhagic manifestations of thrombocythemia in patients with essential thrombocythemia and polycythemia vera: therapeutic implications. J Hematol Thrombotic Diseases.
- Verstovsek S (2014) Ruxolitinib improves over best available therapy in polycythemia vera: RESPONSE study. 80-week follow-up from the RESPONSE trial. EHA 101: 821-829. [Crossref]
- Vykoupil KF, Thiele J, Stangel W, Kromotic E, Georgii A (1980) Polycythemia vera. Histology, ultrastructure and cytogenetics of bone marrow in comparison with secondary polycythemia. Virchows Arch (Path Anat and Histol) 389: 307-324.
- Georgii A, Vykoupil KF, Thiele J (1980) Chronic megakaryocytic granulocytic myelosid-CMGM. Virchows Arch A Path Anat Histol 389: 253-268. [Crossref]
- Adamson JW, Fialkov PF, Murphy S, Prchal JF, Steinman L (1976) Polycythemia vera: stem-cel and probable clonal origin of the disease. N Eng J Med 295: 913-916. [Crossref]
- Fialkov PJ, faguet GB, Jacobson RJ, Vaidya K, Murphy S (1981) Evidence that essential thrombocythemia is a clonal disorder with origin in a multipotent stem cell. Blood 58: 916-919. [Crossref]
- Fialkov PJ, Jacobson RJ, Papayannopoulou T (1977) Chronic myelocytic leukemia: clonal origin in a stem cell common to the granolucyte, erythrocyte, platelet and monocyte/macrophage. Am J Med 63: 125-138. [Crossref]
- Greenberg BR, Wilson FD, Woo L, Jenks HM (1978) Cytogenetics of fibroblasts colonies in Ph-positive chronic myelogenous leukemia. Blood 51: 1039-1044.
- Michiels JJ (1997) Diagnostic criteria of the myeloproliferative disorders (MPD): essential thrombocythemia, polycythemia vera and chronic megakaryocytic granulocytic metaplasia. Neth J Med 51: 57-64. [Crossref]
- Michiels JJ, Kutti J, Bazzan M, Gugliotta L, Marcioli R, et al. (1999) Diagnosis, pathogenesis and treatment of the myeloproliferative disorders essential thrombocythemia, polycythemia vera and essential megakaryocytic granulocytic metaplasia and myelofibrosis. Neth J Med 54: 46-62. [Crossref]
- WHO (2001) Classification of the chronic myeloproliferative diseases (CMPD) polycythemia vera, chronic idiopathic myelofibrosis essential thrombocythemia and cMPD unclassifiable. In: Jaffe SS, Harris NL, Stern A, Vardiman JW (Eds) WHO classification of Tumours of haematopoiesis and lymphoid tissues, Lyon, France. IARC pp: 31-42.
- WHO (2008) Criteria for polycthemia vera, primary myelofibrosis and essential thrombocythemia. In: Swerdlow SH, Campo E, Harris NL (Eds) WHO Classification of Tumours of Haematopoietic and Lympoid Tissues. Lyon France. IARC pp: 40-50.
- Michiels JJ, Kvasnicka HM, Thiele J (2002) Myeloproliferative Disorders: Current perspectives on diagnostic criteria, histopathology and treatment of essential thrombocythemia, polycythemia vera and chronic idiopathic myelofubrosis. Deutschen BibliothekVerlag ME Uwe Grunwald, Much, Germany. ISBN 76: 6-8.
- Michiels JJ, Berneman Z,Schroyens W, De Raeve H (2013) PVSG and WHO vs European Clinical, Molecular andPathological (ECMP) criteria for prefibrotic myeloproliferative neoplasms. World J Hematol 2: 71-88.
- Georgii A (1983) Histopathologie und Klinik der chronischen, myeloproliferatives Erkarnkungen. Verh, Dtsch Ges Path 67: 214-234.
- Burkhardt R, Frisch B, Bartl R (1982) Bone marrowbiopsiese in haematologicaldisorders. J Clin Pathol 8: 257-284. [Crossref]
- Michiels JJ (1981) Erythromelalgia and thrombocythemia. Erasmus University Rotterdam, Thesis Rotterdam.
- Michiels JJ (1997) Erythromelalgia and Thrombocythemia A disease of platelet prostaglandin metabolism. Semin Thromb Hemostas 23: 335-338. [Crossref]
- Kurnick JE, Ward HP, Block MH (1972) Bone marrow sections in the differential diagnosis of polycythemia. Arch Pathol 94:489-499. [Crossref]
- Michiels JJ, van Genderen PJJ, LindemansJ, van Vliet HHDM (1996) Erythromelalgic, thrombotic and hemorrhagic manifestations in 50 cases of thrombocythemia. Leukema Lymphoma 22: 47-56. [Crossref]
- Alvarez-Larran A, Garcia-Pagan JC, Abraldes JG, Arellano E, Reverter JC, el at. (2004) Increased CD11b neutrophil expression in Budd-Chiari syndrome or portal vein thrombosis secondary to polycythaemia vera. Brit J Haematol 124: 329-335. [Crossref]
- Thiele J, Zankovich R, Schneider G, Kremer B, Fischer R, el at. (1988) Primary (essential) thrombocythemia versus polycythemia rubra vera. A histomorphometric analysis of bone marrow features in trephine biopsies. Analyt Quat Cytol Histol 10: 375. [Crossref]
- Thiele J, Kvasnicka HM, Werden C, Zankovich R, Diehl V, el at. (1996) Idiopathic primary osteo-myelofibrosis: a clinico-pathological study on 208 patients with special emphasis on evolution of disease features, differentiation from essential thrombocythemia and variables of prognostic impact. Leukemia Lymphoma 22: 303-317. [Crossref]
- Thiele J, Kvasnicka HM, Diehl V (2005) Initial (latent) polycythemia vera with thrombocytosis mimicking essential thrombocythemia. Acta Haematologica 113: 213. [Crossref]
- Georgii A, Buesche G, Kreft A (1998) The histopathology of chronic myeloproliferative disease. Bailliere's Clin Haematology 11: 721-749. [Crossref]
- Thiele J, Kvasnicka HM, Diehl V, Fischer R, Michiels JJ (1999) Clinicopathological diagnosis and differential criteria of thrombocythemias in various myeloproliferative disorders by histopathology, histochemistry and immunostaining from bone marrow biopsies. Leukemia Lymphoma 33: 207-218. [Crossref]
- Michiels JJ, Thiele J (2002) Clinical and pathological criteria for the diagnosis of essential thrombocythemia, polycythemia vera and idiopathic myelofibrosis (agnogenic myeloid metaplasia). Int J Hematol 76: 133-145. [Crossref]
- Michiels JJ, Juvonen E (1997) Proposal for the revised diagnostic criteria of essential thrombocythemia and polycythemia by the Thrombocythemia Vera Study Group. Semin Thromb Hemostas 23: 339-347. [Crossref]
- Michiels JJ, Barbui T, Finazzi G, Fruchtman SM, Kutti J, et al. (2000) Diagnosis and treatment of polycythemia vera and possible future study designs of the PVSG. Leukemia Lymphoma 36: 239-253. [Crossref]
- Michiels JJ, De Raeve H, Berneman Z, Van Bockstaele D, Hebeda K, et al. (2006) The 2001 World Health Organization (WHO) and updated European clinical and pathological (ECP) criteria for the diagnosis, classification and staging of the Ph1-chromosome negative chronic myeloproliferative disorders (MPD). Semin Thromb Hemostas 32: 307-340. [Crossref]
- Michiels JJ, Berneman Z, Van Bockstaele D, De Raeve H, Schroyens W (2007) Current diagnostic criteria for the chronic myeloproliferative disorders (MPD) essential thrombocythemia (ET), polycythemia vera (PV) and chronic idiopathic myelofibrosis (CIMF). Pathologie Biologie 55: 92-104. [Crossref]
- Michiels JJ, Berneman Z, Schroyens, Hebeda K, Lam K, et al. (2007) WHO bone marrow features and European clinical molecular and pathological criteria for the diagnosis of myeloproliferative disorders. Leukemia Research 31: 1031-1038. [Crossref]
- Tefferi A, Thiele J, Orazi A (2007) Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood 110: 1092-1097. [Crossref]
- Tefferi A, Vardiman JW (2008) Classification and diagnosis of myeloproliferative neoplasms: The 2008 World Health Organization criteria and point of care diagnostic algorithms. Leukemia 22: 14-22. [Crossref]
- Thiele J, Kvasnicka HM, Orazi A, Tefferi A, Birgegard G, et al. (2008) Polycythemia vera, Essential thrombocythaemia and Primary myelofibrosis. In: Swerdlow SH, Campo E, Harris NL (Eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon IARC pp: 40-47.
- Kvasnicka HM, Thiele J (2010) Prodromal myeloproliferative neoplasms: The 2008 WHO classification. Am J Hematol 85: 62-66. [Crossref]
- Thiele J, Kvasnicka HM, Muellauer L, Buxhofer-Ausch V, Gisslinger B, et al. (2011) Essential thrombocythemia versus early primary fibrosis: a multicentre study to validate the WHO classification. Blood 117: 5710-5718. [Crossref]
- Barbui T, Thiele J, Vannucchi AM, Tefferoi A (2013) Problems and pitfalls regarding WHO-defined diagnosis of early/prefibrotic primary myelofibrosis versus essential thrombocythemia. Leukemia [Crossref]
- Michiels JJ, Berneman Z, Schroyens W, De Raeve H (2015) Changing concepts on the diagnostic criteria of myeloproliferative disorders and the molecular etiology and classification of myeloproliferative neoplasms. From Dameshek 1950 to Vainchenker 2005 and beyond. Acta Haematol 133: 36-51. [Crossref]
- Michiels JJ, Valster F, Wielenga J, Schelfout K, De Raeve H (2015) European vs 2015-World Health Organization clinical, molecular andpathological classification of myeloproliferative neoplasms. World J Hematol 6: 16-53.