Abstract
Werner’s syndrome is a rare autosomal recessive premature aging syndrome caused by mutations in the Werner RecQ helicase. Patients typically die in their 5th decade from cardiovascular disease or cancer. There are few reports of the treatment of malignancies in these patients. We previously reported a patient with Werner’s syndrome who expired from multi-organ failure after treatment of AML with intensive chemotherapy. We currently report a patient with Werner’s syndrome and AML who was treated with decitabine, a low intensity regimen commonly used to treat elderly patients. This patient also developed severe toxicity, but recovered and obtained a complete remission. Unfortunately the patient’s disease progressed 5 months later and he then expired.
Key words
Werner’s syndrome, aging, acute myeloid leukemia, chemotherapy, t(9;19)(p13;p13)
Introduction
Werner’s syndrome is a rare autosomal recessive premature aging syndrome caused by mutations in the Werner RecQ helicase, an enzyme involved in repair of double strand DNA breaks and telomere replication [1,2]. When mutated, WRN causes sporadic loss of telomeres, increased DNA damage, genomic instability and premature senescence [3]. The clinical symptoms of Werner’s syndrome begin in the early teens with loss of the growth spurt, followed by more progressive signs and symptoms in the 20’s and 30’s including graying of the hair, hoarseness, scleroderma-like skin changes, bilateral cataracts, type 2 diabetes, hypogonadism, skin ulcers and osteoporosis [1]. Patients typically die in their 5th decade from cardiovascular disease or cancer.
Due to the rarity of this disease; there are very few reports of the treatment of malignancies in patients with Werner’s syndrome. We previously reported a patient Werner’s syndrome who was treated with high dose cytarabine, etoposide and mitoxantrone for newly diagnosed AML [4]. The patient expired from multi-organ failure three weeks after initiation of therapy. We hypothesized that this severe toxicity was secondary to the increased sensitivity of the patient’s normal cells to chemotherapy due to defective DNA repair from Werner’s syndrome. We currently report a second patient with Werner’s syndrome and AML. Based on our prior experience we treated the patient with decitabine, a low intensity regimen commonly used to treat elderly patients. The patient again developed severe toxicity, but this time he recovered and obtained a complete remission. Unfortunately the patient’s disease progressed 5 months later and he then expired.
The patient was 42-year-old man who presented when a low white count was noted on blood work performed prior to surgery for a leg ulcer. The patient had a history of type II diabetes since his early 20’s. He underwent bilateral cataract surgery shortly after that. The patient developed coronary artery disease requiring CABG while in his 30’s. He also had a history of peripheral vascular disease including carotid stenosis, hypercholesterolemia, and hypothyroidism. Recently he had developed a non-healing ulcer on his left heel and was scheduled for surgery. The patient’s medications upon admission were metoprolol, clopidogrel, aspirin, levothyroxine, rosuvastatin, and insulin. Several of the patient’s maternal relatives had cardiovascular disease, but later in life. His father had an unknown type of cancer. No other family members had manifestations of Werner’s syndrome and there was no known consanguinity. The patient was of Hispanic (Puerto Rican) ethnicity.
On physical examination the patient appeared chronically ill and had a distinctive habitus. He was 5 feet tall (several inches shorter than any of his relatives) and weighed 93 pounds. He had a high-pitched, hoarse voice and a beak-like nose. His hair was thin and blonde; however he dyed his hair which had been grey for years. His extremities were particularly thin. His skin was taut with a scleroderma-like appearance. There was a healed ulcer over the right elbow and an open ulcer on the left foot. His feet were flat with calluses on the heels. There were artificial lenses in both eyes. There was a 3/6 systolic murmur to the right of the sternum. There was no hepatosplenomegaly or lymphadenopathy.
Laboratories showed a white blood count (WBC) of 1900/mm3, with 15% neutrophils, 82% lymphocytes, and 3% monocytes. The hemoglobin was 9.7 gm/dl and the platelet count was 191,000/mm3. An echocardiogram demonstrated moderate aortic stenosis. Plain films of the foot demonstrated osteosclerosis and soft tissue calcifications (Figure 1). Osteomyelitis was noted on an MRI of the left foot.
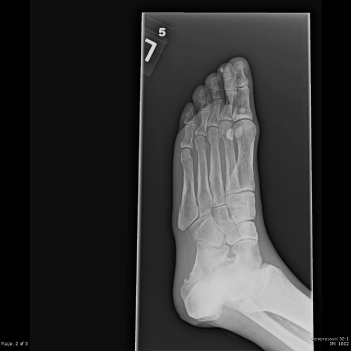
Figure 1. Plain film of the foot demonstrates diffuse osteopenia, osteosclerosis and soft tissue calcifications
The bone marrow aspirate was hemodilute with 16% blasts. The core biopsy was hypercellular with a diffuse increase in reticulin fibrosis. Blasts were estimated at 25% to 30%. On flow cytometry the blasts demonstrated CD13 (subset), CD33 (dim), CD34, CD117, CD11c (dim), HLA-DR, CD64 (dim), CD38, CD71, CD9 (dim), and were negative for CD61, CD56, TdT, and B and T lymphoid markers. Cytogenetics showed 46,XY, t(9;19)(p13;p13)[1]/44,idem,-3,del(5)(q22q33),-7,add(11)(q23),del(17)(p12)[7]/46,XY[12]. FLT3 and NPM were not mutated.
We planned to treat the patient with decitabine 20 mg/m2 daily for 5 days, a low intensity chemotherapy regimen that is commonly used in elderly AML patients. However after the third dose the patient’s course became very similar to that of our previous patient. He developed high fevers, diffuse pulmonary infiltrates, and hypotension requiring vasopressors. Further chemotherapy was withheld. He had progressive respiratory failure and required intubation on day 4 (Figure 2). Bronchoalveolar lavage demonstrated diffuse pulmonary hemorrhage. All cultures showed no growth of organisms. The patient was treated empirically with steroids and broad spectrum antibiotics. Although the patient was critically ill, he gradually improved and was extubated after 18 days of intubation. His renal function which had deteriorated transiently recovered without dialysis. On day 30 his WBC was 5000/mm3 with 60% neutrophils, 29% lymphocytes, 6% monocytes, 1% basophils, 3% metamyelocytes, and 1% myelocytes. The hemoglobin was 9.0 gm/dl and the platelet count was 208,000/mm3. Bone marrow done on day 27 showed a normocellular marrow with trilineage hematopoiesis, mild dyserthropoeisis, dysmegakaryopoiesis and 1% blasts. Cytogenetics demonstrated 46,XY,t(9;19)(p13;p13),del(17)(p12)[13]/46,XY,idem,del(5)(q22q33)[1]/46,XY[9]. Molecular studies performed at the University of Washington School of Medicine (Drs. Junko Oshima and George Martin) through the International Registry of Werner Syndrome demonstrated a homozygous mutation c.1105C>T (p.Arg369Stp) in exon 9. This is the most common WRN mutation among Caucasian patients, seen in 20% of cases [5]. Western analysis showed no detectable WRN protein.
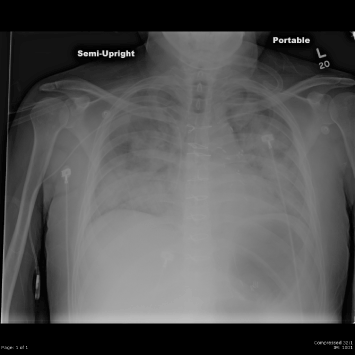
Figure 2. Chest radiograph on day 3 of treatment demonstrates diffuse pulmonary infiltrates
One week later the patient had recovered completely. The patient then received a single day of decitabine, 20 mg/m2 monthly as consolidation. He tolerated this therapy very well. Prior to the third cycle bone marrow morphology and flow cytometry were normal. Cytogenetics showed persistent abnormalities, but with an increasing number of normal metaphases: 46,XY,t(9;19)(p13;p13.3)[7]/ 46,XY[13]. However prior to cycle 5 the patient developed worsening cytopenias and a bone marrow demonstrated 62% blasts. In an attempt to regain the patient’s response he received two days of decitabine 20 mg/m2 for his 5th cycle of therapy. However he developed hypotension, respiratory and renal failure and expired 5 months after his initial diagnosis.
Discussion
Our patient had 4 out of 5 cardinal signs and symptoms of Werner’s syndrome (cataracts, characteristic skin changes, short stature, and premature graying/thinning of the hair) and all of the further signs and symptoms (diabetes mellitus, hypogonadism, osteoporosis, osteosclerosis, soft tissue calcification, premature atherosclerosis, mesenchymal neoplasm, voice changes and flat feet) used by the International Registry of Werner syndrome. The patient did not have known consanguinity. The diagnosis was confirmed by genetic testing.
The hematologic malignancies reported in patients with Werner’s syndrome include acute leukemia, myelodysplastic syndromes, and myelofibrosis [6,7]. These cases are reviewed in our previous report [4]. The presentation is more consistent with that of a hematologic disorder of an elderly patient rather than what is typical for a patient in their 5th decade of life. Patients develop progressive cytopenias and the bone marrow demonstrates dysplasia, fibrosis and complex cytogenetic abnormalities.
We were only able to identify 5 reports of the use of chemotherapy in patients with Werner’s syndrome and acute leukemia, including our previously reported case [4,8-10]. In all cases the outcome was poor, with death due to sepsis, hemorrhage or resistant disease. Tao et al reported a patient with Werner’s syndrome and acute leukemia who obtained a complete remission with transfusions and prednisone [8,9]. This patient subsequently relapsed and received 6-mercaptopurine and vincristine without effect. Other reports include the use of BHAC (N4-behenoyl-1-b-D-arabinofuranosylcytosine) in one patient, BHAC-AMP (aclacinomycin, 6-mercaptopurine, and prednisolone) in another, and DCMP (daunorubicin, cytarabine, 6-mercaptopurine and prednisolone) in a third [10]. Two of the patients died from sepsis, and the third died from leukemic relapse, respectively. Our previous patient expired from multiorgan failure after receiving only 3 days of chemotherapy with cytarabine, etoposide and mitoxantrone4.
It is remarkable that our patient achieved a complete hematologic response after only 3 days of decitabine. In a phase II trial of decitabine in elderly patients with de novo AML where the complete response rate was 24%, the median time from first dose to complete response was 126 days (range 48 to 238 days), equal to 4.5 cycles of therapy [11]. Werner’s cells are hypersensitive to chemotherapy, including DNA cross-linking drugs [12] and topoisomerase inhibitors [13]. It is possible that this hypersensitivity to chemotherapy resulted in both the increased toxicity seen in our patient as well as the rapid response to minimal therapy.
The cytogenetic abnormality, t(9;19)(p13;p13.3) has not been previously reported. Other abnormalities involving chromosome 19 have been reported in AML, especially in patients with acute megakaryoblastic leukemia (FAB-M7). In a report by Dastugue, 11 of 53 patients with acute megakaryoblastic leukemia had abnormalities of chromosome 19, including trisomy 19, loss of 19, add(19)(p13) and t(4;19) [14]. Additionally, Nimer reported frequent gain of chromosome 19 detected by comparative genomic hybridization that could not be identified by g-banding in patients with acute megakaryoblastic leukemia [15]. Our patient’s bone marrow demonstrated some findings commonly seen in patients with acute megakaryoblastic leukemia including dysplasia and fibrosis, however flow cytometry did not support a megakaryoblastic lineage.
Conclusion
This patient with Werner’s syndrome had an initial response to decitabine, but still had significant toxicity. He tolerated a very low dose of decitabine well for consolidation but this dose failed to treat his leukemia effectively.
Acknowledgements
2021 Copyright OAT. All rights reserv
The authors wish to thank Drs. Junko Oshima and George Martin for genetic testing and the Irving Hansen Foundation for ongoing support.
This work was supported by a grant from the Irving Hansen Foundation.
References
- Werner CWO (1904) Uber Katarakt in Verbindung mit Sclerodermie. (Doctoral Dissertation, Kiel University). Schmidt and Klaunig, Kiel.
- Huang S, Li B, Gray MD, Oshima J, Mian IS, et al. (1998) The premature ageing syndrome protein, WRN, is a 3’-5’ exonuclease. Nat Genet 20: 114-116. [Crossref]
- Crabbe L, Jauch A, Naeger CM, Holtgreve-Grez H, Karlseder J (2007) Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc Natl Acad Sci U S A 104: 2205-2210. [Crossref]
- Seiter K, Qureshi A, Liu D, Galvin-Parton P, Arshad M, et al. (2005) Severe toxicity following induction chemotherapy for acute myelogenous leukemia in a patient with Werner’s syndrome. Leuk Lymphoma 47: 1091-1095. [Crossref]
- Friedrich K, Lee L, Leistritz DF, Nürnberg G, Saha B, et al. (2010) WRN mutations in Werner syndrome patients: genomic rearrangements, unusual intronic mutations and ethnic-specific alterations. Hum Genet 2010; 128: 103-111. [Crossref]
- Bjornberg Alf (1976) Werner’s syndrome and malignancy. Acta Dermatovener (Stockholm) 56: 149-150.
- Sato K, Goto M, Nishioka K, Arima K, Hori N, et al. (1988) Werner's syndrome associated with malignancies: five case reports with a survey of case histories in Japan. Gerontology 34: 212-218. [Crossref]
- Tao LC, Stecker E, Gardner HA (1971) Werner’s syndrome and acute myeloid leukemia. Can Med J 105: 951-968. [Crossref]
- Stecker E, Gardner HA (1970) Werner’s syndrome. Lancet 2: 1317. [Crossref]
- Mita M, Ishibashi T, Shichishima T, Maruyama Y (1996) Myelodysplastic syndrome with multiple chromosome aberrations in a patient with Werner’s syndrome. Rinsho Ketsueki 37: 725-730. [Crossref]
- Cashen AF, Schiller GJ, O’Donnell MR, DiPersio JF (2010) Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J Clin Oncol 28: 556-561. [Crossref]
- Poot M, Yom JS, Whang SH, Kato JT, Gollahon KA, et al. (2001) Werner syndrome cells are sensitive to DNA cross-linking drugs. FASEB J 15: 1224-1226. [Crossref]
- Pichierri P, Franchitto A, Mosesso P, Proietti de Santis L, et al. (2000) Werner’s syndrome lymphoblastoid cells are hypersensitive to topoisomerase II inhibitors in the G2 phase of the cell cycle. Mutat Res 459: 123-133. [Crossref]
- Dastugue N, Lafage-Pochitaloff M, Pagès MP, Radford I, Bastard C, et al. (2002) Cytogenetic profile of childhood and adult megakaryoblastic leukemia (M7): a study of the Groupe Francais de Cytogenetique Hematologique (GFCH). Blood 100: 618-626. [Crossref]
- Nimer SD, MacGrogan D, Jhanwar S, Alvarez S. (2002) Chromosome 19 abnormalities are commonly seen in AML, M7. Blood 100: 3838. [Crossref]