Abstract
Classically, molecular chaperones play a pivotal role in the maintenance of cellular proteostasis and thus, in the safeguarding of the cell homeostasis while reducing the deleterious effects of extracellular and intracellular stresses. They are also active players in immunologically relevant scenarios such as the activation of innate immunity, antitumour immunity, and autoimmune diseases. It is currently accepted that misdirected immune responses may target self-antigens and generate severe inflammatory responses, a typical signature of autoimmune diseases. In addition to numerous components in immune responses, chaperone proteins are also detected in the extracellular fluids and have been implicated in autoimmune and inflammatory diseases acting as pro- and anti-inflammatory factors. In several inflammatory pathologies, chaperones are greatly induced as a direct consequence of the inflammatory stress and are released from the cell thanks to a poorly understood mechanism. These extracellular chaperones are capable to stimulate anti-inflammatory regulatory T cell responses, thereby inducing the negative feedback control of inflammation. Therefore, it has been proposed that immunization with heat-shock protein peptides could prevent the development of certain diseases. In this article we review the basics of the stress response, summarize current controversies over the role of extracellular chaperones in inflammatory reactions and autoimmunity, and discuss the cytoprotective and immunoregulatory roles of heat-shock proteins, a challenging subject that may open a new avenue for the drug discovery and treatment of diseases related to autoimmune disturbs.
Introduction to the stress response
Cells are always exposed to a number of sudden and potentially harmful variations of their biological milieu. With the purpose to protect proteins from misfolding, denaturation and/or aggregation, cells trigger a fast response characterized by a number of events able to protect them from the hostile environment, and restore a balanced and safe new steady-state of protein homeostasis commonly referred to as ‘proteostasis’ [1,2]. Importantly, such response builds a physiological network that shields cells and tissues from the risky challenges they may encounter, such as heat, cold, toxics, chemicals, UV light, radiation, drugs, infections, infestations, inflammation, pH variations, osmotic changes, nutrient deprivation, oxidative stress, hypoxia-ischemia, apoptotic stimuli, and stressful conditions for individuals such as psychiatric disorders and socially traumatic experiences [3]. The generation of a new proteostasis network implies an immediate role in protein synthesis, folding, disaggregation, or degradation, processes that encompass the translational machinery, molecular chaperones and their associated cochaperones, the ubiquitin-proteasome machinery, and the autophagy system.
The stress response to heat-shock was originally described in the early ‘60s by the Italian researcher Ferruccio Ritossa [4]. One of his colleagues accidentally switched the temperature of his incubator where Drosophila cells were grown, and a quite different puffing pattern was acquired by those cells. The biological consequence was an exceptional increase of transcriptional activity for a family of proteins that, for this particular reason, were generically named heat-shock proteins (HSPs). Then, it was noted that HSPs are expressed in a highly conserved manner in all organisms from bacteria to humans, to the point that many times they may be efficiently interchanged. HSPs form oligomeric complexes constitutively expressed under normal conditions (ranging up 5-10% of the total protein). They function as molecular chaperone machineries capable to regulate the folding of all proteins, intracellular transport of soluble and particulate cargoes among the different subcellular compartments, repair or degradation of proteins, refolding of misfolded proteins, etc. [5]. In addition to being constitutively expressed, HSPs are also greatly induced by the onset of stress (up to 15%). It should be noted that the terms HSP and chaperone are frequently used as synonyms. However, while all HSPs are always chaperones, not all chaperones are induced by heat-shock.
The biological function of molecular chaperones is not limited to solve abnormal situations, but they also have an active participation in the biological function of native factors such as steroid receptors, protein-kinases, transcription factors, enzymes, oncogenes, structural proteins, etc. Actually, the 90-kDa heat-shock protein, Hsp90, has more than four hundred client proteins [6].
As it was stated above, those molecular chaperones induced by heat stress are also called HSPs. However, temperature is not the sole stimulus to trigger the stress response, but also a wide variety of other stimuli (UV radiation, chemicals, toxic compounds, metals, inappropriate pH or osmotic pressure, nutrient starvation, oxidants, fever, cancer, infections, neurodegenerative diseases, etc [7]). Also, the heat-shock response can come in handy during the normal growth cycle of the cells even without the existence of stressors in the medium.
The essential role of the heat-shock factor
The HSP genes are regulated by a master transcription factor, the HSF (Heat-Shock Factor) [2]. In vertebrates four HSF members have evolved (HSF1, HSF2, HSF3, and HSF4), the HSF1 being so far the most abundant and best characterized member of the family. The inactive monomer of HSF1 is cytoplasmic and forms oligomers with the Hsp90-based heterocomplex [8]. Upon the exposure of cells to stress, HSF1 homotrimerizes, translocates to the nucleus, and acquires specific DNA-binding and transcription-enhancing activities [9,10]. How stressing situations are sensed by the cell and conveyed to HSF1 is not entirely understood. As mentioned before, a great number of stresses can trigger a heat-shock response, therefore it has been postulated that different types of molecular sensors may exist for each type of stimulus. Alternatively, it could be that a given proteotoxic environment may converge in a common signal able to initiate the biological response. The classical model postulates that under normal conditions, the association of molecular chaperones to HSF1 maintains this transcription factor inactive. When cells are subjected to stressing situations, the increased level of protein misfolding would release the Hsp90-based complex from HSF1 monomers allowing its trimerization [8,11]. Other models propose that HSF1 has itself the ability to detect proteotoxic conditions, as evidenced by the capacity of purified HSF1 to homotrimerize in vitro upon the onset of different types of stresses [12].
The induction of the stress response results in high levels of expression of molecular chaperones, which bind to nascent chains and other metastable soluble proteins preventing misfolding and/or aggregation. During the attenuation of stress and the subsequent recovery steps, the pool of non-native proteins dissipates and protein homeostasis is therefore restored. However the persistence of the original problem may perpetuate mechanisms responsible for proteostasis maintenance by constant activation of influencers. This is manifested with the existence of a steady-state in pathologies such as Alzheimer’s disease, chronic emotional distress, psychiatric diseases, cancer, and diabetes [1].
Molecular chaperones
The molecular chaperone family comprises a very heterogeneous group of proteins that may be grouped into eight subfamilies [1,13,14]: 1)-The small HSPs/a-crystalin group, which show molecular weight ranging from 12-kDa to 43-kDa. There are 10 mammalian sHSPs, all of them are cytosolic and most of which contain an α-crystalline domain [15]. They favour the client protein refolding by Hsp70 by forming large homo-oligomeric cages able to trap misfolded proteins and preventing the formation of undesirable intra-or intermolecular interactions. This process is ATP-independent and thought to be complementary for those commanded by other ATP-dependent chaperones. 2) The DNAJ/Hsp40 subfamily, characterized by the presence of a J domain that permits interactions with Hsp70. These chaperones can be thought of as adaptors that provide versatility to Hsp70 function [16] assisting protein refolding by presenting misfolded proteins to Hsp70 and by enhancing its ATPase activity. 3)- The Hsp60 mitochondrial chaperonin-like protein is essential for maturation and maintenance of the mitochondrial proteome and is therefore intimately linked to energy production [13]. 4)- The Hsc70/Hsp70 subfamily of chaperones with intrinsic ATPasa activity that is enhanced by Hsp40. It works in a cooperative fashion with Hsp90 in several heterocomplexes, and is also recruited to newly synthesized polypeptides by the ribosome-associated complex [17]. Importantly, Hsp70 shows antiapoptotic properties [18]. 5) The Hsp90 scaffold protein is able to form large heterocomplexes with other chaperones and cochaperones. It is perhaps the most versatile protein of the entire family and certainly is the most abundant soluble protein of the cell [19]. 6)- The large HSPs/Clp subfamily of AAA+ ATPases (molecular weight of 100-kDa to 110-kDa), which have the ability to solubilise almost any protein that becomes aggregated after severe stress, but are not required under normal growth conditions [20]. They work associated with Hsp70 extracting trapped polypeptides from aggregates via a threading mechanism. 7)- An additional group comprises chaperones present in the endoplasmic reticulum that participates in the proper folding and eventual degradation of new born proteins, such as the glucose regulating proteins (Grp), the ubiquitine and lectin chaperones calnexin and calreticulin. 8)- Immunophilis, endogenous proteins that are characterized by the presence of a signature domain named PPIase (peptidyl-prolyl-(cis/trans)-isomerase), i.e., an enzymatic activity of isomerase able to interconvert reversibly Xaa-Pro bonds High molecular weight immunophilins are frequently bound to Hsp90 playing the role of cochaperones or forming a functional regulatory unit with the chaperone [21,22].
The Hsp90-Hsp70-Immunophilin complex
Both chaperones Hsp90 and Hsp70 are highly expressed in all types of cells, and they cooperate in most of the complexes with client proteins. The cytoplasmic pool of Hsp90 forms homodimers, each monomer unit consisting of three domains: a) The N-terminal domain contains an ATP binding site whose hydrolysis to ADP is critical in the chaperoning activity of the Hsp90 homodimer. b) The M- domain is a flexible middle region able to interact with a great numbers of client proteins and is linked to the N-terminal domain by a highly charged linker sequence. The intrinsic ATPase activity of Hsp90 requires an arginine located in the “catalytic loop” of the middle domain [23]. c) The C-terminal domain is responsible for the dimerization of the chaperone and also possesses a key pentapeptide MEEVD sequence that permits the association of TPR-domain proteins such as immunophilins, Hop, WISp39, Tom70, CHIP, Tah1, etc. [19,24,25].
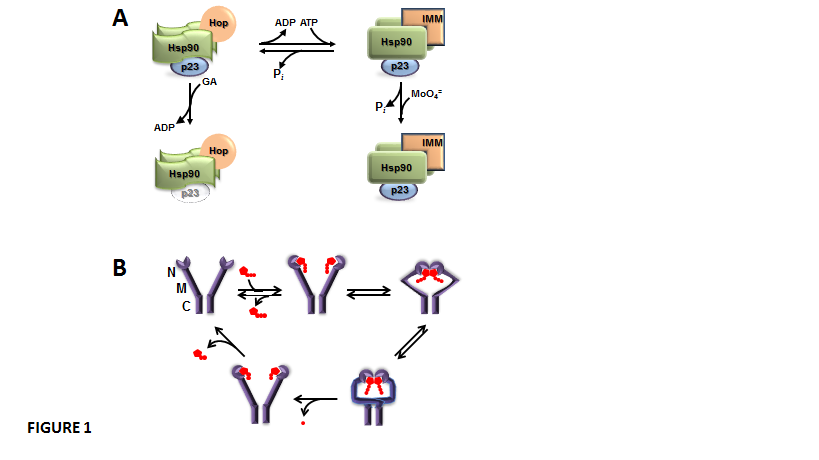
Dimers of Hsp90 are in dynamic equilibrium between two states, the ADP-bound isoform and the ATP-bound isoform, which are able to form different oligomeric heterocomplexes with other chaperones (Figure 1-A). The ADP-bound Hsp90 shows higher affinity for the TPR-domain-containing cochaperone Hop. Hop brings together Hsp70 and Hsp90. Hop also prevents Hsp90 from effectively chaperoning client proteins whose binding favours a high, sustained level of ATP [26,27]. Hop binding to Hsp90 blocks ATP binding and the ATPase activity of the chaperone, which makes binding of the cochaperone p23 significantly weak or void [28,29]. This ADP-bound conformation of Hsp90 is favoured by the Hsp90 disrupting agent geldanamycin, a benzoquinone ansamycin antibiotic that shows high affinity for the nucleotide binding site of the chaperone. In this state, the associations of both p23 and TPR-domain immunophilins to the complex are impaired. On the other hand, when ADP is exchanged with ATP, the ATP-bound isoform of Hsp90 is stabilized by p23 binding, and other TPR proteins such as the high molecular weight immunophilins are efficiently recruited [26,27]. The ATP-bound isoform of Hsp90 is a better modulator of the biological activity of client proteins. It appears that p23 binding to Hsp90 changes the nature of ATP binding to Hsp90 and prevents the intrinsic ATPase activity of the chaperone [30], which in turn may affect the biological activity of the Hsp90-bound client factor. Hsp90 interacts with hundreds of client proteins and other signaling molecules, including steroid receptors, p53, tyrosine-kinases, oncogenes, Stat5, NOS, HSF1, Akt, Chk1, Mdm2, IkB kinases, cytoskeletal proteins, importins, nucleoporins, histones, DNA helicases, telomerase reverse transcriptase, DNA polymerases, SmyD methyltransferases, p300 acetyltransferase, HDAC6, Aha1, SGT1, and Cdc37. In other words, Hsp90 creates a broad scaffold platform for the regulation of signalling cascades, nuclear architecture and cell function [28,31,32]. The mechanism involved in client protein-Hsp90 interaction also involves the ATPase intrinsic activy of the chaperone. Thus, Hsp90 exists in a conformational equilibrium between two states ¾an open (V-shaped) conformation and a close conformation (Figure 1-B), a state where ATP-hydrolysis takes place, the N-terminal domains dissociate, and Hsp90 returns to the open conformation again [33].
Perhaps one of the most remarkable biological functions of high molecular weight immunophilins associated to Hsp90/Hsp70 complexes is the ability to interact with the dynein/dynactin motor complex [34], a property used to favour the retrotransport of soluble factors in an active manner using cytoskeletal tracks. This machinery is used for the transport of proteins such as GR [35], MR [36], ecdysone receptor [37], RAC3/AIF complexes [38], NF-kB [39], etc., as well as for viral particles when cells are infected by a virus [40]. Importantly, Hsp90-based complexes have been related to microbial invasion of host cells [41,42] and increased antigenicity of pathogen soluble factors [43].
Role of heat-shock proteins in immunity
The best biological incentive to trigger the innate immunity response is the exposure to a foreign molecule in a hazardous milieu. The critical decision of an organism for responding or ignoring that stimulus is taken by innate immune recognition receptors upon their activation by PAMP ( pathogen-associated molecular patterns) molecules, i.e. factors associated with groups of pathogens that are small molecular motifs conserved within a class of pathogens (for example lipopolysaccharides, bacterial CpG DNA, etc.) and recognized by cells of the innate immune system by PRRs (or Pattern Recognition Receptors) [44]. A number of these PRRs have been cloned and showed to belong to the expanding family of toll-like receptors (TLRs). An alternative pathway for the activation of innate immunity is that proposed by the ‘danger theory’ [45]. According to this hypothesis, the innate immunity can be additionally activated by endogenous substances released by the damaged or stressed tissue itself. In this way, stressed cells can messenger stress to other cells of any type. Some of the stress signals released by cells are the HSPs induced in response to the insult, such that they are potential candidates for signalling tissue damage or cellular stress. In this regard, some of them such as Hsp60 and Hsp70 have been actually found capable of signalling through CD14, TLR-2, and TLR-4 [46,47].
The first findings reported a heat-shock-like protein described as a glia-axon transfer protein of the squid giant axon [48], as well as the fact that Hsp70 is released from cells by a mechanism not impaired by inhibitors of the known secretory pathways [49]. This original early observation was deemed not relevant, and the presence of HSPs in the extracellular environment was disregarded for many years until it was demonstrated that recombinant Hsp70 activates cells of the immune system [50]. These findings were disputed based on the possibility that the activation of immune cells was due to contamination [51,52]. Nonetheless, these worries were ruled out after experiments using pure recombinant Hsp70 isolated from insect cells, non-recombinant Hsp70, treatments with antibiotics, boiling treatments, incubation in serum free media, etc. [53,54]. Today, it is well established and broadly accepted that Hsp70 is responsible for the activation of macrophages, monocytes, dendritic cells, natural killer cells, hepatocytes, etc. [55-58]. Moreover, extracellular HSPs have been shown to work as potent immunostimulatory or immunosuppressive molecules depending on the circumstances by which they interact with cells [59].
In addition to Hsp70, other HSPs have also been found in the extracellular milieu, such as Hsp60 [60], Hsp90 [61-63], Grp78/BIP [64,65], and Hsp27 [66]. The biological relevance of these extracellular factors has been reinforced by the detection of Hsp70 in the serum of patients suffering from a number of pathologies such as chronic inflammation [67], myocardial infarction [68], lung injury [69], coronary artery disease [70,71], infections [67], cancer [72,73], ischemia/reperfusion events [74], diabetes [75], hypertension during pregnancy [76], etc. HSPs can also be detected in the serum of healthy individuals, although at lower levels [59]. Importantly, the presence of Hsp70 in plasma is correlated with improved survival of critically ill patients [77,78]. Other extracellular HSPs such as Hsp27 [66], Hsp60 [79], and Hsp90 [61,80] have also been linked to several diseases such as pancreatic carcinoma, coronary heart disease, cancer metastasis or systemic lupus erythematosus.
Heat-shock protein secretion
Regarding the origin of extracellular HSPs, while it was originally demonstrated a basal HSP release from healthy cells [49], others researchers proposed that the source of extracellular HSPs could be the consequence of cell lysis after necrosis [81]. Later on it was demonstrated that HSPs are released by an active mechanism independent of cell death [82]. Nevertheless, these studies cannot preclude the possibility that necrosis could be an additional source of extracellular HSPs, so two different sources of extracellular HSPs can coexist ¾the active pathway due to a non-conventional secretory mechanism, and a passive one secondary to cell death and subsequent lysis [55,83].
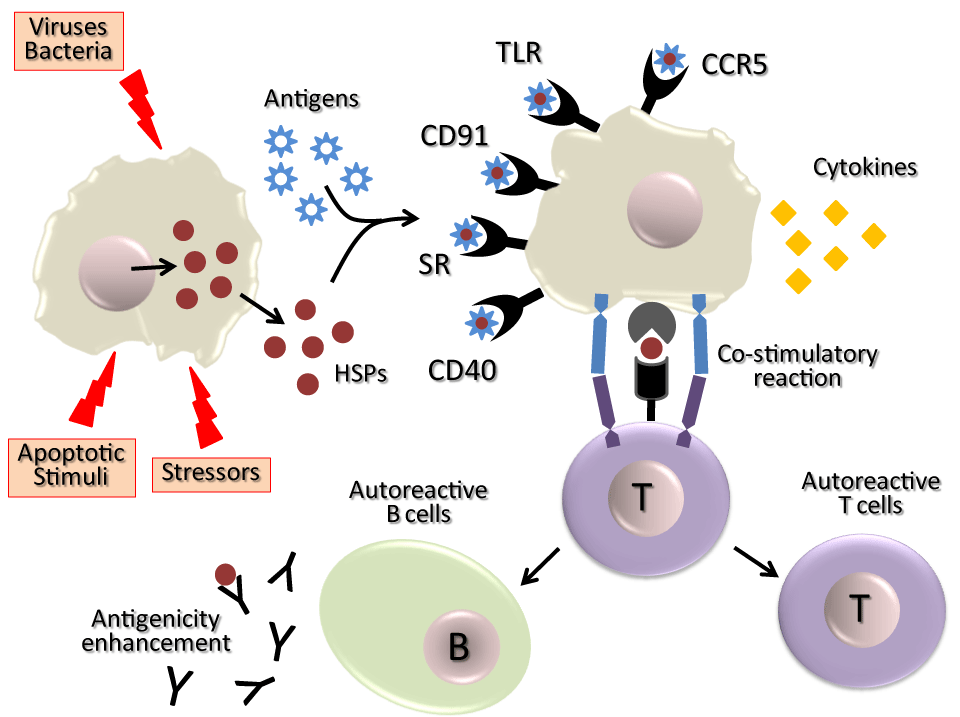
Following HSP release from external pathogens, foreign cells, autologous inflamed or necrotizing tissue (such as tumours or those target tissues of autoimmune diseases), they are recognized by receptors expressed in the surface of immune cells of the host ¾TLR-4, TLR-4 like, TLR-2, CD14, CD91, CD94, LOX-1, SR-A, or as an immunodominant antigens [55,84,85] 8–11. With the contribution of these released HSPs, the immune system of the host is therefore “informed” about the presence of pathologic processes (Figure 2).
The secretion mechanism of Hsp70 is perhaps the most studied, although definitive conclusions are still uncertain. An active mechanism for Hsp70 secretion has not be demonstrated to date, whereas typical inhibitors of the classic endoplasmic reticulum-Golgi pathway, such as brefaldin-A, showed no effect on the release of Hsp70 [49]. On the other hand, this chaperone does not possess a consensus peptide signal for secretion via the classic ER-Golgi pathway. Therefore, it is likely that the export mechanism is via a putative alternative pathway called the non-classical or unconventional secretory pathway [86], which could also be shared by other factors such as some interleukins, galectins, and growth factors, although unifying common steps for this proposed mechanism remain still unrevealed [86]. Three possible scenarios for Hsp70 active secretion could be its export a)- via the lysosome-endosome pathway, in which Hsp70 is translocated into the lysosome lumen via ATP-binding cassette (ABC) transport-like system and further transported outside cells via the endocytic process [87]; b)- via secretory-like granules loaded of Hsp70 in similar manner as to those granules used by exocrine cells and the endocrine and neuroendocrine systems to release hormones and granines in secretory vesicles [88]; and c)- via extracellular vesicles [55] derived from the plasma membrane by membrane blebbing, ectosome vesicles, and/or by endocytosis, i.e., due to the formation of multiple vesicles with the same topology as the plasma membrane (exosomes) [89]. Regardless of the origin of the vesicles, other HSPs such as Hsp90, Hsp60, Hsp27, Grp78/BIP have also been detected in the lumen and membrane of these extracellular vesicles [90]. It is thought that the HSPs in the lumen of the vesicle are the consequence of protein being trapped during the formation of the vesicle since HSPs are very abundant in the cytosol. Therefore, it is expected that vesicles should burst to release the intravesicular HSPs allowing them to interact with other proteins and cells. Alternatively, vesicles can fuse with the membranes of cells, releasing the lumen content into the cytosol as well as those HSP associated with the vesicle membrane, as reported for Hsp70 [53,91] and Hsp60 [60,92]. HSPs are highly concentrated in the vesicles (for example, in the mM order for Hsp70 [55]), which favours interactions with other factors and cell surface receptors increasing the biological activity of the chaperone. It has been reported that the specific activity of Hsp70 in these vesicles is more than 250 fold greater for activating macrophages than the soluble Hsp70 [55]. Similar to Hsp70, Hsp90 also lacks a secretory signal; therefore it has been postulated as being exported via exosomes [93,94] or by direct translocation across the plasma membrane [95].
Other molecular chaperones are also present in the external cell surface, such as Grp78/BIP [96], Grp94 on the plasma membrane of sarcoma cells [97], Hsp60 on the surface of human endothelial cells [98] and T cell-lines [99], as well as in mouse liver and spleen [100]. Hsp90, which was first detected on the cell surface of tumour cells [101], was subsequently found on monocytes [43], mesenchymal cells [102], and neuroblastoma cells [61,103].
Role of extracellular chaperones in immunity
Exported HSPs trapped the attention of several laboratories because of their potential action on the immune system, where which they function as signalling molecules. Accordingly, it has been stated that extracellular Hsp70 is able to activate monocytes, macrophages natural killer and dendritic cells [55]. Also, Hsp70 is a stimulating factor for autoimmunity [104,105] by a peptide-specific immune response that involves the a2-macroglobulin receptors CD91, CD40 and OLR1/LOX-1 expressed in antigen-presenting cells [106]. The innate immunity response is triggered by recognition of toll-like receptor (TLR) signals, one of the activating signals being the exported HSPs. Thus, to evaluate if exported Hsp70 shows antigenic properties, isolated lymphocytes from rat spleens were exposed to purified Hsp70, and the result was lymphocyte proliferation [107]. Then, it was demonstrated that the immunogenic form of Hsp70 corresponds to a well-conserved 14 amino acid sequence (amino acids 139–153) [108].
Additionally, HSPs can also play a role in generating antigen-specific T cell responses [109] It is thought that complexed peptides with Hsp70, Hsp90B1/gp96, calreticulin, etc. are delivered to antigen-presenting cells by receptor-mediated internalization of the HSPs. This could make them available for further processing and presentation to the major histocompatibility complex. Inasmuch as specific receptor-mediated mechanisms exist for binding and internalization of HSPs [110], it may be envisaged that the cross-presentation of HSP-derived antigenic determinants is a realistic mechanism for cross-priming by antigen-presenting cells. Consequently, it is logical to propose that these HSPs highly induced in several types of pathologies are indeed targets of the adaptive immune response.
It has been demonstarted that exported Hsp70 increases neutrophil chemotaxis and their microbicidal ability [111], as well as macrophage phagocytosis properties [112], and modulation of the response of monocytes to endotoxin [113]. Similarly, extracellular Hsp70 is related to both immunostimulatory and immunosuppressive activities [114]. Nonetheless, the exact molecular mechanisms responsible for such modulation of the response of cells of the immune system are still poorly understood. It was reported that during the inflammatory response generated by tumours, both IFN-γ and IL-10 induce the active release of both Hsp70 isoforms Hsc73 and Hsp72) from the malignant tissue [115]. It is still uncertain whether these secreted HSPs produce antitumour immunogenicity responses.
Taking advantage of the capability of HSPs to target a chaperoned antigen to antigen-presenting cells and elicit cross-presentation, it has been developed immunotherapy strategies using the secretable forms of HSPs. Thus, it is known that the Hsp90B1/gp96 secreted from tumour cells carries an antigenic peptide able to activate specific antitumour cytotoxic T lymphocyte responses [116]. This property is also shared by exported Hsp70 and Hsp90 [117]. The endoplasmic reticulum chaperone BiP (Binding of immunoglobulin protein) is also exported from tumour tissues and capable to elicit antigen-specific tumour immunity [118]. This secreted BiP complexed with antigenic peptides is taken up by dendritic cells and cross- presented in association with MHC class I molecules. This leads to cytotoxic T lymphocyte responses, which in turn stimulates tumour cells to produce their own endogenous “cellular vaccine”. Such immunogenic strategy could be more advantageous than a peptide-generated vaccine approach because single peptide-based cancer vaccines have certain disadvantages. For example, single peptide vaccination usually induces a HLA-restricted cytotoxic T lymphocyte response, thereby allowing tumour cells to escape from lymphocyte recognition. Contrarily, because HSPs are associated to a broad-spectrum antigenic peptide repertoire, it is likely that the subsequent induction of a cytotoxic T lymphocyte response is more effective because it is directed against multiple antigens. In addition, this type of secreted HSP-based cancer vaccine may be useful for all classes of patients regardless of HLA restrictions. In short, gene modification of HSPs for secretion could be a practical strategy to provide an exceptional beneficial approach for cancer immunotherapy.
Similar to cytoplasmic chaperones, it has been proposed that Hsp90B1/gp6, an endoplasmic reticulum-resident partner of the chaperone Hsp90, might participate in mechanisms that are critical for cell growth, differentiation, and responses to endoplasmic reticulum stress [119]. Also, it is a natural adjuvant for chaperoning antigenic self peptides into the immune surveillance pathways, and may also be involved in the maintenance of morphostasis and self tolerance. Recently, it was demonstrated that high levels of up-regulation of Hsp90B1/gp96 in regenerating liver and thymus are followed by signs of transient autoimmunity, augmented apoptosis in thymus, and the presence of autoreactive NKT and regulatory T cells that might be involved in the control of rapid liver growth induced by partial hepatectomy [120].
Hsp90 is a key regulator of innate immunity
It is accepted that exogenous antigens are processed by two main pathways [83,121]: a)- the classical MHC class I loading pathway involving the transporter-associated antigen-presenting (TAP)-dependent pathway, and b)- the endosome-recycling pathway, which involves a post-Golgi loading mechanism of MHC class I in endocytic compartments. It has been demonstrated that soluble Hsp90 is able to translocate extracellular antigens from endosome to cytosol for TAP-dependent cross-presentation [122], where antigens are processed in the endosomes by cathepsin S and other peptidases, and thereafter are loaded in endocytic compartments onto MHC class I molecules. These complexes are recycled from plasma membranes [123,124]. Hsp90 also works as an efficient route-finder for those chaperoned antigens targeted to the cross-presentation pathway [125,126]. Also, it has recently been demonstrated that Hsp90-cancer antigen peptide complexes are efficiently cross-presented by monocyte-derived dendritic cells and stimulated peptide-specific cytotoxic T lymphocyte responses [127]. Also, it was demonstrated [128] that stimulation of CD4+T cells by dendritic cells DC that had been activated by Hsp90-peptide complexes involved pathways that share many of the essentials mechanisms involved in stimulation of antigen cross presentation.
Hsp90 empowers the chaperoned ligands to activate an immune response. It is well known that unmethylated single-stranded DNA containing a cytosine–phosphate–guanine (CpG) motif binds TLR9 receptors of plasmacytoid dendritic cells (pDCs) and B cells, which results in the production of IFN-α [129]. However, these cells normally do not respond to self-DNA, a phenomenon that was explained by the fact that both bacterial and viral DNA have multiple CpG nucleotides able to bind and consequently activate TLR9 more efficiently than mammalian self-DNA contains, which contains a lower number of these motifs and are usually masked by methylation. There are situations where self-DNA might have the potential to activate TLR9, but it may not succeed since it fails to access the TLR9-containing endolysosomal compartments [130]. Synthetic CpG-DNAs have been assayed¾ CpG-A, able to stimulate the receptors of pDC cells and produce IFN-α, and CpG-B, which is inactive in this regard [130], but stimulates the release of IL-6 and TNF-α in these cells. While CpG-A forms large multimeric aggregates and is retained for long periods of time in early endosomes, whereas CpG-B is a monomer, cannot oligomerize, and rapidly translocate from early to late endosomes or lysosomes of pDCs [131]. Such a long-lasting retention of CpG-A results in prolonged activation of TLR9 signaling, leading to strong IFN-α production. In other words, the retention time of the CpG/ TLR9 complex in endosomes is a key determinant for the TLR9-dependent signalling cascade. Inasmuch as Hsp90 binds and retains client ligands within early endosomes, studies specifically conducted to evaluate the role of such interaction demonstrated that human pDCs pulse Hsp90·CpG-A complexes inducing a larger production of IFN-α than the monomeric CpG-A [130]. In contrast to human dendritic cells, murine cells also express TLR7 receptor in addition to TLR9 [132]. In this system, the Hsp90·CpG-A complex is retained within early endosomes for longer periods leading to sustained activation of murine cells and eliciting TLR9 signaling for IFN-α production, which is indeed TLR9-mediated since TLR9 KO cells do not produce IFN-α. By contrast, CpG-A alone was transfered to late endosomes and lysosomes. Importantly, not only CpG-A·Hsp90 complexes, but also CpG-B·Hsp90 complexes are strong stimulating ligands of TLR9 resulting in high production of IFN-α.
It may be stated that extracellular Hsp90 has the capacity to chaperone CpGs in early endosomes leading to a more abundant production IFN-α. Therefore, there are two important considerations derived from that property: a)- extracellular Hsp90 (and HSPs in general) could play a crucial role in the pathogenesis of nucleic acid-mediated autoimmune diseases, for example in systemic lupus erythematosus; and b)- it may be implied that the use of extracellular HSPs in combination with CpG may enhance the biological effectiveness of cancer vaccines.
Heat-shock proteins and autoimmune diseases
The participation of HSPs in the pathogenesis of autoimmune diseases has been analyzed using two basic animal models: a) - artificially induced arthritis by the natural saturated terpenoid alkane pristane, adjuvant-induced arthritis, avridine-induced arthritis, arthritis generated by Streptococcus cell wall and collagen type II [133]; and b)- insulin-dependent diabetes mellitus model in non-obese diabetic mice (NOD)[134]. The adjuvant-induced arthritis is generated by intracutaneous injection with heat-killed Mycobacterium tuberculosis. Arthritis can be transferred from diseased injected animals to healthy naive animals with a single T cell clone specific for a particular sequence of Hsp60 M. tuberculosis isoform. Thus, the microbial agent initiates a self-perpetuating autoimmune process in a susceptible individual generating antibodies against the host’s Hsp60, and perhaps an epitope of the cartilage proteoglycan [135]. Regarding the potential advantages of this phenomenon from the perspective of therapeutic applications, it was observed the induction of tolerance after immunization with the HSP. Accordingly, pristine oil-generated arthritis disease shows high levels of IL-2 and IFN-γ after in vitro restimulation of arthritic mice splenocytes with mycobacterial Hsp60 [136].Therefore, mycobacterial Hsp60 is capable of preimmunize mice and protected them from pristane actions.
Similarly, anti-Hsp70 antibodies have been found in the sera of patients with malaria [137]. HSPs have been shown to play a cardinal role in antigen direct presentation and cross-presentation leading to CD8+ T cell activation [109]. There is strong evidence to support the notion that cytosolic HSPs such as Hsp70 and Hsp90, and those of the endopasmic reticulum such as Hsp90B1/gp96, have the ability to bind antigenic peptides generated within the cells, as well as they are also part of the endogenous pathway of antigen presentation via the major histocompatibility complex class I molecules [138,139]. Peptides that are chaperoned by HSPs when released extracellularly, or HSP-peptide complexes assambled in vitro, are taken up by antigen-presenting cells such as dendritic cells via α2-macroglobulin receptor (CD91)-mediated endocytosis, resulting in representation by the major histocompatibility complex molecules [138,140]. Vaccination of mice with Hsp70, Hsp90 and Hsp90B1/gp96 isolated from murine tumour cells elicits immune response sufficient for tumour rejection and suppression of metastatic tumour progression [141]. This tumour immunity was shown to result from tumour-derived peptides associated with the HSPs, rather than from the HSPs themselves [117].
Insulin-dependent diabetes mellitus is the other example of Hsp60 involvement in autoimmune diseases. The regular disease is the clinical consequence of pancreas β-cell destruction by T cells. Infection of non-obese diabetic mice with M. avium prevents the development of diabetes [142]. Similarly immunization of NOD mice with mycobacterial Hsp60 significantly reduces the incidence of the disease [143], suggesting that autoantibodies against Hsp60 could play a major role in the etiology of this type of diabetes.
Systemic lupus erythematosus is other prototype of autoimmune disease characterized by the presence of plasma autoantibodies against self-nuclear structures, such as dsDNA and RNA-containing antigens, including Sm and RNP [144,145]. This disease is characterized by the presence of autoantibodies against Hsp90 [146] and enhanced expression of Hsp90 in peripheral blood mononuclear cells of patients [147]. Cytosolic Hsp90 translocates to the nuclear compartment under stressful conditions, which raises the possibility that Hsp90 may bind self-DNA in the nucleus. In a following step, self-DNA associated with endogenous Hsp90 can be released into the extracellular milieu when cells undergo necrosis, triggering the production of IFN-α by denditric cells. Serum Hsp90α levels has been found increased in patients with lupus [148,149], and these high concentrations are significantly decreased after treatment. Moreover, immunodepletion of extracellular Hsp90 from serum decreased IFN-α production by pDCs [148], indicating that depletion of Hsp90 might induce remission and prevent end-organ damage. Thus, control of deregulated pDC activation and IFN-α production represents an attractive treatment strategy for systemic lupus erythematosus.
Collectively, extracellular Hsp90 plays a crucial role in the disease activity of SLE and that Hsp90 inhibitors have promise for the treatment of IFN-α-mediated autoimmune diseases. In line with this postulate, it has recently been reported the evaluation of small molecule inhibitors of Hsp90 as potential therapeutic target for this disease in an autoimmune mouse model [150]. The novel resorcinolic triazolone inhibitor STA9090/ganetespib [151] was found to be equally efficacious and tolerable when compared to an effective weekly dosing regimen of the standard-of-care immunosuppressive agent cyclophosphamide. Importantly, co-treatment of ganetespib with a suboptimal, intermittent dosing schedule of cyclophosphamide resulted in superior therapeutic indices and maximal disease control. These findings highlight the potential of HSP90 inhibition as an alternative, and potentially complementary, strategy for therapeutic intervention in systemic lupus erythematosus.
HSPs in the Autoimmune Diseases of the central nervous system (CNS)
The very first reports of HSPs release from neuronal cells were related with the secretion of both Hsp70 and Hsp110 along with actin [49]. Further publications reported that Hsp70 is released from glial cells and taken up by adjacent neurons [48,152]. It has been demonstrated that many neurons have reduced expression of some HSPs due to inadequate HSP gene transcription activity [153]. However glial cells adjacent to synapses release HSPs into the extracellular medium and chaperones are captured by neuronal receptors [152,154,155]. The functions of such non-neuronal HSPs are not properly understood to date, but probably they may provide some chaperoning properties to neurons and protection from programmed cell death, which can accompany neurotransmission [155].
In view of the limited regenerative capacity of CNS, protection against immune responses requires special mechanisms of self defence. This is achieved due to singular conditions able to reduce the exposure to the immune system, referred to as ‘immune privilege’ [156]. This is possible due to the existence of blood-brain barrier, the limited expression of major histocompatibility complex molecules of the brain, and neuronal expression of molecules with immunosuppressive properties, e.g. TGF-β and CD200. These characteristics contribute to protect neurons from microglia and T-cell-induced damage. However activated immune cells can invade the CNS under specific conditions through selective receptor-ligand interactions [157], and the list of neurological diseases where T cells are involved in targeting neurons or their axons, directly or indirectly, is continuously growing [158]. The most frequent diseases where the immune system targets neurons are multiple sclerosis, the paraneoplastic cerebellar degeneration, and Parkinson’s disease.
Multiple sclerosis is a multifocal inflammatory autoimmune disease affecting only the CNS in which the target is the myelin and the myelin-producing cells, oligodendrocytes. This disease may be considered a standard model of an autoimmune disease of the CNS from the perspective of the molecular mechanism of action of HSPs. Myelin is destroyed in patches, resulting in the formation of astrocytic scars (or plaques), showing high accumulation of T cells and macrophages, increased concentrations of cytokines and chemokines, and antibodies to myelin proteins [159]. Although the exact etiology of the disease is not fully understood, current evidence suggests a critical role for the immune system in its pathogenesis. The inflammatory and oxidative environment generated in the CNS of these patients favours the overexpression of most of HSPs, including Hsp70 [160,161], whose induction occurs predominantly in oligodendrocytes [162]. The biological consequences of Hsp70 induction are dual. Based on their activity as chaperone, the release of Hsp70 into the milieu act as an adjuvant that binds immunogenic peptides, which increases the inflammatory local reaction and autoimmunity. On the other hand, Hsp70 is an antiapoptotic factor [18,163,164] and, as all chaperones do, it prevents the accumulation of abnormal protein aggregates [165], a common histopathological hallmark that contributes to neuronal degeneration [166]. Therefore, inadequate induction of Hsp70 can increase the rate of protein aggregation upon the onset of stressing situations leading to apoptosis, a typical end for most neurodegenerative and autoimmune diseases such as multiple sclerosis. Accordingly, it has been reported that Hsp70 overexpression in neuronal cell cultures and also mouse models of neurodegenerative diseases shows advantageous actions to ameliorate the severity of the phenotypes by reducing the number and size of inclusion bodies and the accumulation of disease-causing factors [161].
Since Hsp70 works as a neuroprotective factor for ischemia-induced cell death [167], it may be argued that the overexpression of the chaperone in multiple sclerosis lesions might protect the CNS against the typical inflammatory environment of this disease [168]. In line with this, it has been demonstrated that the stimulating action of proinflammatory cytokines (IL-1, TNFa, IFN-g, etc.) in glial cells induce Hsp70 expression, which in turn exerts favourable final effects on glial cells and neurons [162]. However, all coins have two sides. At the same time, exported Hsp70 leads to the activation of the NF-kB transcription factor due to the activation of TLRs; this induces the production of proinflammatory cytokines (IL-12, IL-1b, IL-6, TNF-a, and GM-CSF) [50,81,135], chemokine secretion (MCP-1, RANTES, and MIP-1a) [169], and nitric oxide production [170] by macrophages and dendritic cells. Moreover, extracellular Hsp70 enhances the expression of CD83, CD86, CD40 and the major histocompatibility complex class II on dendritic cells, as well as the migration of these cells to draining lymph nodes [81,171]. All of these elicit priming adaptive immune responses. Therefore, it is thought that the acute stress induces the release of Hsp70, which works as a danger signal to prepare the immune system against a possible pathogen challenge.
Taken all together the above exposed observations together it can be concluded that the inflammatory phase occurring at the initial stages of multiple sclerosis is a sort of preconditioning stimulus for glial cells to induce Hsp70 expression and release, which in turn has a protective action on neurons in the subsequent neurodegenerative phase. This permits to envisage an obvious scenario ¾a failure to induce Hsp70 or the insufficient production of HSPs in the CNS is a possible determining factor for multiple sclerosis development, and conducts to postulate therapeutic strategies focusing on HSP up-regulation for different neuropathologies. This logical hypothesis, however, cannot be applied for the case of multiple sclerosis because there is an additional conflicting factor ¾the strong autoimmune reaction that accompanies multiple sclerosis triggers a process of neurodegenerative damage that leads to the typical clinical signs of the disease. Thus, extracellular Hsp70 is actually a harmful rather than beneficial factor. Compared to other CNS pathologies such as Alzheimer’s and Parkinson’s diseases, the released Hsp70 in multiple sclerosis strongly exacerbates the immune response by acting as a very efficient adjuvant for myelin peptides and as a proinflammatory cytokine [172]. These effects cannot be counterbalanced by Hsp70 beneficial actions such as the reduction of protein misfolding or antiapoptotic effects. These concepts are particularly valid for viral infections. Researchers are considering the possibility that viruses, and also bacterial infections, may cause multiple sclerosis. Viruses are known to cause inflammation and a breakdown of myelin (or demyelination). Therefore, it is highly possible that a virus could trigger the disease. Several viruses and bacteria are currently being investigated to determine if they are involved in the development of multiple sclerosis. These studies include measles virus, human herpes virus-6 (HHV-6), and Epstein-Barr virus (EBV).
All these features can also been observed in autoimmune diseases such as glaucomatous pathology, where elevated serum titers of autoantibodies to many optic nerve components [173] and retina antigens [174] are present, as well as abnormal T-cell subsets [175]. In this case the master chaperoned involved in the disease is Hsp27. In addition to the intrinsic neuroprotective functions of Hsp27, like other HSPs, this chaperone also serves as an antigenic stimulus activating the innate and adaptive immune response during glaucomatous neurodegeneration [176]. By serving as a danger signal, up-regulation of Hsp27 may facilitate detection and elimination of stressed retinal ganglion cells by the immune system. Thus, uncontrolled immune activity, including T-cell mediated cytotoxicity to retinal ganglion cells and their axons, may eventually facilitate the progression of neurodegeneration. In summary, the overall summary of these biological events tells us that potential errors during the processing of the native cellular interactions are relevant for the conversion of protective immunity and self-limiting inflammatory responses into chronic neurodegeneration and autoimmune diseases.
Concluding remarks
The export of HSPs is still a poorly understood process as a consequence of our reduced understanding of the actual mechanism for the release of the chaperones and the lack of a standard action in that extracellular milieu, with biological consequences that are even opposite to the classical and broadly accepted actions of the these proteins. One piece of evidence, however, is clear today after so many years of controversy: the extracellular HSPs are not artefacts. They play roles as extracellular factors ranging from ligands to activate cell surface receptors to favour antigenicity of pathogenic factors, such that autoimmune responses are developed. The overall function of these chaperones seems to be at the level of signalling or cellular communication rather than at the traditional chaperone activity. In this regard, to date no overwhelming experimental evidence exist that allow us to affirm that extracellular HSPs play the same chaperone activity outside cells, to the point that the requirement of co-chaperones and other factors associated to them for playing their intracellular roles are unlikely not required for their extracellular functions as signalling molecules. A particularly relevant effect is the modulation of the immune system, which is visualized as a novel priming event for cellular communication under stressing situations. Ultimately, these exported molecular chaperones represent a new opportunity to explore eventual therapeutic approaches to modulate the biological response of exported HSPs in the development and progression of autoimmune diseases.
Acknowledgement
The costs of this publication were supported by grants from the Agencia Nacional de Promoción Científica y Tecnológica (PICT 2014-3433), Universidad de Buenos Aires (UBACYT 20020130100318BA) and Instituto Nacional del Cáncer (INC2015)
References
- Labbadia J, Morimoto RI (2015) The biology of proteostasis in aging and disease. Annu Rev Biochem 84: 435-464. [Crossref]
- Anckar J, Sistonen L (2011) Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem 80: 1089-1115. [Crossref]
- Christians ES, Benjamin IJ (2006) Heat shock response: lessons from mouse knockouts. Handb Exp Pharmacol 139-152. [Crossref]
- Ritossa F (1962) A new puffing pattern induced by temperature shock and DNP in Drosophila. Experientia 18: 571-573.
- Hendrick JP, Hartl FU (1993) Molecular chaperone functions of heat-shock proteins. Annu Rev Biochem 62: 349-384 [Crossref]
- Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, et al. (2012) Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150: 987-1001. [Crossref]
- Ciocca DR, Arrigo AP, Calderwood SK (2013) Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: an update. Arch Toxicol 87: 19-48. [Crossref]
- Guo Y, Guettouche T, Fenna M, Boellmann F, Pratt WB, et al. (2001) Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J Biol Chem 276: 45791-45799. [Crossref]
- Voellmy R, Boellmann F (2007) Chaperone regulation of the heat shock protein response. Adv Exp Med Biol 594: 89-99. [Crossref]
- Hensold JO, Hunt CR, Calderwood SK, Housman DE, Kingston RE (1990) DNA binding of heat shock factor to the heat shock element is insufficient for transcriptional activation in murine erythroleukemia cells. Mol Cell Biol 10: 1600-1608. [Crossref]
- Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, et al. (2007) HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev 21: 2172-2181. [Crossref]
- Ahn SG, Thiele DJ (2003) Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev 17: 516-528. [Crossref]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU (2013) Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem 82: 323-355. [Crossref]
- Kampinga HH1 (2006) Chaperones in preventing protein denaturation in living cells and protecting against cellular stress. Handb Exp Pharmacol 1-42. [Crossref]
- Haslbeck M, Franzmann T, Weinfurtner D, Buchner J (2005) Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol 12: 842-846. [Crossref]
- Kampinga HH, Craig EA (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 11: 579-592. [Crossref]
- Preissler S, Deuerling E (2012) Ribosome-associated chaperones as key players in proteostasis. Trends Biochem Sci 37: 274-283. [Crossref]
- Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, et al. (2001) Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol 3: 839-843. [Crossref]
- Erlejman AG, Lagadari M, Toneatto J, Piwien-Pilipuk G, Galigniana MD (2014) Regulatory role of the 90-kDa-heat-shock protein (Hsp90) and associated factors on gene expression. Biochim Biophys Acta 1839: 71-87. [Crossref]
- Bukau B, Weissman J, Horwich A (2006) Molecular chaperones and protein quality control. Cell 125: 443-451. [Crossref]
- Erlejman AG, Lagadari M, Galigniana MD (2013) Hsp90-binding immunophilins as a potential new platform for drug treatment. Future Med Chem 5: 591-607. [Crossref]
- Erlejman AG, Lagadari M, Harris DC, Cox MB, Galigniana MD1 (2014) Molecular chaperone activity and biological regulatory actions of the TPR-domain immunophilins FKBP51 and FKBP52. Curr Protein Pept Sci 15: 205-215. [Crossref]
- Cunningham CN, Southworth DR, Krukenberg KA, Agard DA (2012) The conserved arginine 380 of Hsp90 is not a catalytic residue, but stabilizes the closed conformation required for ATP hydrolysis. Protein Sci 21: 1162-1171. [Crossref]
- Quintá HR, Galigniana NM, Erlejman AG, Lagadari M, Piwien-Pilipuk G, et al. (2011) Management of cytoskeleton architecture by molecular chaperones and immunophilins. Cell Signal 23: 1907-1920. [Crossref]
- Picard D (2002) Heat-shock protein 90, a chaperone for folding and regulation. Cell Mol Life Sci 59: 1640-1648. [Crossref]
- Sullivan WP, Owen BA, Toft DO (2002) The influence of ATP and p23 on the conformation of hsp90. J Biol Chem 277: 45942-45948. [Crossref]
- Morishima Y, Kanelakis KC, Murphy PJ, Shewach DS, Pratt WB (2001) Evidence for iterative ratcheting of receptor-bound hsp70 between its ATP and ADP conformations during assembly of glucocorticoid receptor.hsp90 heterocomplexes. Biochemistry 40: 1109-1116. [Crossref]
- Pratt WB, Toft DO (2003) Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 228: 111-133. [Crossref]
- Kanelakis KC, Murphy PJ, Galigniana MD, Morishima Y, Takayama S, et al. (2000) hsp70 interacting protein Hip does not affect glucocorticoid receptor folding by the hsp90-based chaperone machinery except to oppose the effect of BAG-1. Biochemistry 39: 14314-14321. [Crossref]
- Karagöz GE, Duarte AM, Ippel H, Uetrecht C, Sinnige T, et al. (2011) N-terminal domain of human Hsp90 triggers binding to the cochaperone p23. Proc Natl Acad Sci U S A 108: 580-585. [Crossref]
- Galigniana MD, Echeverria PC, Erlejman AG, Piwien-Pilipuk G (2010) Role of molecular chaperones and TPR-domain proteins in the cytoplasmic transport of steroid receptors and their passage through the nuclear pore. Nucleus 1: 299-308. [Crossref]
- Pratt WB, Galigniana MD, Morishima Y, Murphy PJ (2004) Role of molecular chaperones in steroid receptor action. Essays Biochem 40: 41-58. [Crossref]
- Hessling M, Richter K, Buchner J (2009) Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nat Struct Mol Biol 16: 287-293. [Crossref]
- Harrell JM, Kurek I, Breiman A, Radanyi C, Renoir JM, et al. (2002) All of the protein interactions that link steroid receptor.hsp90.immunophilin heterocomplexes to cytoplasmic dynein are common to plant and animal cells. Biochemistry 41: 5581-5587. [Crossref]
- Galigniana MD, Radanyi C, Renoir JM, Housley PR, Pratt WB (2001) Evidence that the peptidylprolyl isomerase domain of the hsp90-binding immunophilin FKBP52 is involved in both dynein interaction and glucocorticoid receptor movement to the nucleus. J Biol Chem 276: 14884-14889. [Crossref]
- Galigniana MD, Erlejman AG, Monte M, Gomez-Sanchez C, Piwien-Pilipuk G (2010) The hsp90-FKBP52 complex links the mineralocorticoid receptor to motor proteins and persists bound to the receptor in early nuclear events. Mol Cell Biol 30: 1285-1298. [Crossref]
- Vafopoulou X, Steel CG (2012) Cytoplasmic travels of the ecdysteroid receptor in target cells: pathways for both genomic and non-genomic actions. Front Endocrinol (Lausanne) 3: 43. [Crossref]
- Colo GP, Rubio MF, Nojek IM, Werbajh SE, Echeverría PC, et al. (2008) The p160 nuclear receptor co-activator RAC3 exerts an anti-apoptotic role through a cytoplasmatic action. Oncogene 27: 2430-2444. [Crossref]
- Erlejman AG, De Leo SA, Mazaira GI, Molinari AM, Camisay MF, et al. (2014) NF-κB transcriptional activity is modulated by FK506-binding proteins FKBP51 and FKBP52: a role for peptidyl-prolyl isomerase activity. J Biol Chem 289: 26263-26276. [Crossref]
- Zhong L, Qing K, Si Y, Chen L, Tan M, et al. (2004) Heat-shock treatment-mediated increase in transduction by recombinant adeno-associated virus 2 vectors is independent of the cellular heat-shock protein 90. J Biol Chem 279: 12714-12723. [Crossref]
- 41. Khan NA, Shin S, Chung JW, Kim KJ, Elliott S, et al. (2003) Outer membrane protein A and cytotoxic necrotizing factor-1 use diverse signaling mechanisms for Escherichia coli K1 invasion of human brain microvascular endothelial cells. Microb Pathog 35: 35-42. [Crossref]
- Dhakal BK, Mulvey MA (2009) Uropathogenic Escherichia coli invades host cells via an HDAC6-modulated microtubule-dependent pathway. J Biol Chem 284: 446-454. [Crossref]
- 43. Cecchini P, Tavano R, Polverino de Laureto P, Franzoso S, Mazzon C, et al. (2011) The soluble recombinant Neisseria meningitidis adhesin NadA(Delta351-405) stimulates human monocytes by binding to extracellular Hsp90. PLoS One 6: e25089.
- Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20: 197-216. [Crossref]
- Matzinger P (1998) An innate sense of danger. Semin Immunol 10: 399-415. [Crossref]
- Asea A (2003) Chaperokine-induced signal transduction pathways. Exerc Immunol Rev 9: 25-33. [Crossref]
- Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, et al. (2001) Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem 276: 31332-31339. [Crossref]
- Tytell M, Greenberg SG, Lasek RJ (1986) Heat shock-like protein is transferred from glia to axon. Brain Res 363: 161-164. [Crossref]
- Hightower LE, Guidon PT Jr (1989) Selective release from cultured mammalian cells of heat-shock (stress) proteins that resemble glia-axon transfer proteins. J Cell Physiol 138: 257-266. [Crossref]
- Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, et al. (2000) HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med 6: 435-442. [Crossref]
- Bendz H, Marincek BC, Momburg F, Ellwart JW, Issels RD, et al. (2008) Calcium signaling in dendritic cells by human or mycobacterial Hsp70 is caused by contamination and is not required for Hsp70-mediated enhancement of cross-presentation. J Biol Chem 283: 26477-26483. [Crossref]
- Gao B, Tsan MF (2003) Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor alpha release by murine macrophages. J Biol Chem 278: 174-179. [Crossref]
- Vega VL, Rodriguez-Silva M, Frey T, Gehrmann M, Diaz JC, et al. (2008) Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J Immunol 180: 4299-4307. [Crossref]
- Zheng H, Nagaraja GM, Kaur P, Asea EE, Asea A (2010) Chaperokine function of recombinant Hsp72 produced in insect cells using a baculovirus expression system is retained. J Biol Chem 285: 349-356. [Crossref]
- De Maio A (2011) Extracellular heat shock proteins, cellular export vesicles, and the Stress Observation System: a form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to Ferruccio Ritossa. Cell Stress Chaperones 16: 235-249. [Crossref]
- Specht HM, Ahrens N, Blankenstein C, Duell T, Fietkau R, et al. (2015) Heat Shock Protein 70 (Hsp70) Peptide Activated Natural Killer (NK) Cells for the Treatment of Patients with Non-Small Cell Lung Cancer (NSCLC) after Radiochemotherapy (RCTx) - From Preclinical Studies to a Clinical Phase II Trial. Front Immunol 6: 162. [Crossref]
- Calderwood SK, Gong J (2016) Heat Shock Proteins Promote Cancer: It's a Protection Racket. Trends Biochem Sci 41: 311-323. [Crossref]
- Gaipl US, Multhoff G, Scheithauer H, Lauber K, Hehlgans S, et al. (2014) Kill and spread the word: stimulation of antitumor immune responses in the context of radiotherapy. Immunotherapy 6: 597-610. [Crossref]
- Pockley AG, Multhoff G (2008) Cell stress proteins in extracellular fluids: friend or foe? Novartis Found Symp 291: 86-95. [Crossref]
- Merendino AM, Bucchieri F, Campanella C, Marcianò V, Ribbene A, et al. (2010) Hsp60 is actively secreted by human tumor cells. PLoS One 5: e9247. [Crossref]
- Tsutsumi S, Neckers L (2007) Extracellular heat shock protein 90: a role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci 98: 1536-1539. [Crossref]
- Eustace BK, Jay DG (2004) Extracellular roles for the molecular chaperone, hsp90. Cell Cycle 3: 1098-1100. [Crossref]
- Picard D (2004) Hsp90 invades the outside. Nat Cell Biol 6: 479-480. [Crossref]
- Kern J, Untergasser G, Zenzmaier C, Sarg B, Gastl G, et al. (2009) GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood 114: 3960-3967. [Crossref]
- Delpino A, Castelli M (2002) The 78 kDa glucose-regulated protein (GRP78/BIP) is expressed on the cell membrane, is released into cell culture medium and is also present in human peripheral circulation. Biosci Rep 22: 407-420. [Crossref]
- Liao WC, Wu MS, Wang HP, Tien YW, Lin JT (2009) Serum heat shock protein 27 is increased in chronic pancreatitis and pancreatic carcinoma. Pancreas 38: 422-426. [Crossref]
- Njemini R, Lambert M, Demanet C, Mets T (2003) Elevated serum heat-shock protein 70 levels in patients with acute infection: use of an optimized enzyme-linked immunosorbent assay. Scand J Immunol 58: 664-669. [Crossref]
- Dybdahl B, Slordahl SA, Waage A, Kierulf P, Espevik T, et al. (2005) Myocardial ischaemia and the inflammatory response: release of heat shock protein 70 after myocardial infarction. Heart 91: 299-304. [Crossref]
- Ganter MT, Ware LB, Howard M, Roux J, Gartland B, et al. (2006) Extracellular heat shock protein 72 is a marker of the stress protein response in acute lung injury. Am J Physiol Lung Cell Mol Physiol 291: L354-361. [Crossref]
- Zhu J, Quyyumi AA, Wu H, Csako G, Rott D, et al. (2003) Increased serum levels of heat shock protein 70 are associated with low risk of coronary artery disease. Arterioscler Thromb Vasc Biol 23: 1055-1059. [Crossref]
- Zhang X, Xu Z, Zhou L, Chen Y, He M, et al. (2010) Plasma levels of Hsp70 and anti-Hsp70 antibody predict risk of acute coronary syndrome. Cell Stress Chaperones 15: 675-686. [Crossref]
- Azuma K, Shichijo S, Takedatsu H, Komatsu N, Sawamizu H, et al. (2003) Heat shock cognate protein 70 encodes antigenic epitopes recognised by HLA-B4601-restricted cytotoxic T lymphocytes from cancer patients. Br J Cancer 89: 1079-1085. [Crossref]
- Faure O, Graff-Dubois S, Bretaudeau L, Derre L, Gross DA, et al. (2004) Inducible Hsp70 as target of anticancer immunotherapy: Identification of HLA-A*0201-restricted epitopes. Int J Cancer 108: 863-870. [Crossref]
- Hecker JG, McGarvey M (2011) Heat shock proteins as biomarkers for the rapid detection of brain and spinal cord ischemia: a review and comparison to other methods of detection in thoracic aneurysm repair. Cell Stress Chaperones 16: 119-131. [Crossref]
- Oglesbee MJ, Herdman AV, Passmore GG, Hoffman WH (2005) Diabetic ketoacidosis increases extracellular levels of the major inducible 70-kDa heat shock protein. Clin Biochem 38: 900-904. [Crossref]
- Molvarec A, Prohaszka Z, Nagy B, Szalay J, Fust G, et al. (2006) Association of elevated serum heat-shock protein 70 concentration with transient hypertension of pregnancy, preeclampsia and superimposed preeclampsia: a case-control study. J Hum Hypertens 20: 780-786. [Crossref]
- Ziegler TR, Ogden LG, Singleton KD, Luo M, Fernandez-Estivariz C, et al. (2005) Parenteral glutamine increases serum heat shock protein 70 in critically ill patients. Intensive Care Med 31: 1079-1086. [Crossref]
- Pittet JF, Lee H, Morabito D, Howard MB, Welch WJ, et al. (2002) Serum levels of Hsp 72 measured early after trauma correlate with survival. J Trauma 52: 611-617. [Crossref]
- Zhang X, He M, Cheng L, Chen Y, Zhou L, et al. (2008) Elevated heat shock protein 60 levels are associated with higher risk of coronary heart disease in Chinese. Circulation 118: 2687-2693. [Crossref]
- Shimp SK, 3rd, Chafin CB, Regna NL, Hammond SE, Read MA, et al. (2012) Heat shock protein 90 inhibition by 17-DMAG lessens disease in the MRL/lpr mouse model of systemic lupus erythematosus. Cell Mol Immunol 9: 255-266. [Crossref]
- Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK (2000) Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol 12: 1539-1546. [Crossref]
- Hunter-Lavin C, Davies EL, Bacelar MM, Marshall MJ, Andrew SM, et al. (2004) Hsp70 release from peripheral blood mononuclear cells. Biochem Biophys Res Commun 324: 511-517. [Crossref]
- Tamura Y, Yoneda A, Takei N, Sawada K1 (2016) Spatiotemporal Regulation of Hsp90-Ligand Complex Leads to Immune Activation. Front Immunol 7: 201. [Crossref]
- Quintana FJ, Cohen IR (2005) Heat shock proteins as endogenous adjuvants in sterile and septic inflammation. J Immunol 175: 2777-2782. [Crossref]
- Ohashi K, Burkart V, Flohé S, Kolb H (2000) Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol 164: 558-561. [Crossref]
- Nickel W, Seedorf M (2008) Unconventional mechanisms of protein transport to the cell surface of eukaryotic cells. Annu Rev Cell Dev Biol 24: 287-308. [Crossref]
- Mambula SS, Calderwood SK (2006) Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J Immunol 177: 7849-7857. [Crossref]
- Evdonin AL, Martynova MG, Bystrova OA, Guzhova IV, Margulis BA, et al. (2006) The release of Hsp70 from A431 carcinoma cells is mediated by secretory-like granules. Eur J Cell Biol 85: 443-455. [Crossref]
- Théry C, Ostrowski M, Segura E (2009) Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 9: 581-593. [Crossref]
- De Maio A, Vazquez D (2013) Extracellular heat shock proteins: a new location, a new function. Shock 40: 239-246. [Crossref]
- Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, et al. (2010) Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest 120: 457-471. [Crossref]
- Gupta S, Knowlton AA (2007) HSP60 trafficking in adult cardiac myocytes: role of the exosomal pathway. Am J Physiol Heart Circ Physiol 292: H3052-3056. [Crossref]
- Clayton A, Turkes A, Navabi H, Mason MD, Tabi Z (2005) Induction of heat shock proteins in B-cell exosomes. J Cell Sci 118: 3631-3638. [Crossref]
- McCready J, Sims JD, Chan D, Jay DG (2010) Secretion of extracellular hsp90alpha via exosomes increases cancer cell motility: a role for plasminogen activation. BMC Cancer 10: 294. [Crossref]
- Li W, Sahu D, Tsen F (2012) Secreted heat shock protein-90 (Hsp90) in wound healing and cancer. Biochim Biophys Acta 1823: 730-741. [Crossref]
- Zhang Y, Liu R, Ni M, Gill P, Lee AS (2010) Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem 285: 15065-15075. [Crossref]
- Altmeyer A, Maki RG, Feldweg AM, Heike M, Protopopov VP, et al. (1996) Tumor-specific cell surface expression of the-KDEL containing, endoplasmic reticular heat shock protein gp96. Int J Cancer 69: 340-349. [Crossref]
- Pfister G, Stroh CM, Perschinka H, Kind M, Knoflach M, et al. (2005) Detection of HSP60 on the membrane surface of stressed human endothelial cells by atomic force and confocal microscopy. J Cell Sci 118: 1587-1594. [Crossref]
- Soltys BJ, Gupta RS (1997) Cell surface localization of the 60 kDa heat shock chaperonin protein (hsp60) in mammalian cells. Cell Biol Int 21: 315-320. [Crossref]
- Belles C, Kuhl A, Nosheny R, Carding SR (1999) Plasma membrane expression of heat shock protein 60 in vivo in response to infection. Infect Immun 67: 4191-4200. [Crossref]
- Ferrarini M, Heltai S, Zocchi MR, Rugarli C (1992) Unusual expression and localization of heat-shock proteins in human tumor cells. Int J Cancer 51: 613-619. [Crossref]
- Gronthos S, Zannettino AC, Graves SE, Ohta S, Hay SJ, et al. (1999) Differential cell surface expression of the STRO-1 and alkaline phosphatase antigens on discrete developmental stages in primary cultures of human bone cells. J Bone Miner Res 14: 47-56. [Crossref]
- Cid C, Regidor I, Poveda PD, Alcazar A (2009) Expression of heat shock protein 90 at the cell surface in human neuroblastoma cells. Cell Stress Chaperones 14: 321-327. [Crossref]
- Multhoff G (2006) Heat shock proteins in immunity. Handb Exp Pharmacol : 279-304. [Crossref]
- Rodríguez-Iturbe B, Pons H, Quiroz Y, Lanaspa MA3, Johnson RJ3 (2014) Autoimmunity in the pathogenesis of hypertension. Nat Rev Nephrol 10: 56-62. [Crossref]
- Todryk SM, Gough MJ, Pockley AG (2003) Facets of heat shock protein 70 show immunotherapeutic potential. Immunology 110: 1-9. [Crossref]
- Gomes KA, Stupka JA, Diana A, Parra GI (2008) [Molecular characterization of calicivirus strains detected in outbreaks of gastroenteritis occurring in Argentina during 2005 and 2006]. Rev Argent Microbiol 40: 222-228. [Crossref]
- Wendling U, Paul L, van der Zee R, Prakken B, Singh M, et al. (2000) A conserved mycobacterial heat shock protein (hsp) 70 sequence prevents adjuvant arthritis upon nasal administration and induces IL-10-producing T cells that cross-react with the mammalian self-hsp70 homologue. J Immunol 164: 2711-2717. [Crossref]
- Srivastava P (2002) Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol 2: 185-194. [Crossref]
- Arnold-Schild D, Hanau D, Spehner D, Schmid C, Rammensee HG, et al. (1999) Cutting edge: receptor-mediated endocytosis of heat shock proteins by professional antigen-presenting cells. J Immunol 162: 3757-3760. [Crossref]
- Ortega E, Hinchado MD, Martin-Cordero L, Asea A (2009) The effect of stress-inducible extracellular Hsp72 on human neutrophil chemotaxis: a role during acute intense exercise. Stress 12: 240-249. [Crossref]
- Wang R, Kovalchin JT, Muhlenkamp P, Chandawarkar RY (2006) Exogenous heat shock protein 70 binds macrophage lipid raft microdomain and stimulates phagocytosis, processing, and MHC-II presentation of antigens. Blood 107: 1636-1642. [Crossref]
- Abboud PA, Lahni PM, Page K, Giuliano JS Jr, Harmon K, et al. (2008) The role of endogenously produced extracellular hsp72 in mononuclear cell reprogramming. Shock 30: 285-292. [Crossref]
- Pockley AG, Muthana M, Calderwood SK (2008) The dual immunoregulatory roles of stress proteins. Trends Biochem Sci 33: 71-79. [Crossref]
- Barreto A, Gonzalez JM, Kabingu E, Asea A, Fiorentino S (2003) Stress-induced release of HSC70 from human tumors. Cell Immunol 222: 97-104. [Crossref]
- Yamazaki K, Nguyen T, Podack ER (1999) Cutting edge: tumor secreted heat shock-fusion protein elicits CD8 cells for rejection. J Immunol 163: 5178-5182. [Crossref]
- Udono H, Srivastava PK (1994) Comparison of tumor-specific immunogenicities of stress-induced proteins gp96, hsp90, and hsp70. J Immunol 152: 5398-5403. [Crossref]
- Tamura Y, Hirohashi Y, Kutomi G, Nakanishi K, Kamiguchi K, et al. (2011) Tumor-produced secreted form of binding of immunoglobulin protein elicits antigen-specific tumor immunity. J Immunol 186: 4325-4330. [Crossref]
- Staron M, Yang Y, Liu B, Li J, Shen Y, et al. (2010) gp96, an endoplasmic reticulum master chaperone for integrins and Toll-like receptors, selectively regulates early T and B lymphopoiesis. Blood 115: 2380-2390. [Crossref]
- Radosevic-Stasic B, Jakovac H, Grebic D, Trobonjaca Z, Mrakovcic-Sutic I, et al. (2012) Heat shock protein Gp96 as potential regulator of morphostasis after partial hepatectomy in mice. Curr Aging Sci 5: 254-262. [Crossref]
- Kutomi G, Tamura Y, Okuya K, Yamamoto T, Hirohashi Y, et al. (2009) Targeting to static endosome is required for efficient cross-presentation of endoplasmic reticulum-resident oxygen-regulated protein 150-peptide complexes. J Immunol 183: 5861-5869. [Crossref]
- Imai T, Kato Y, Kajiwara C, Mizukami S, Ishige I, et al. (2011) Heat shock protein 90 (HSP90) contributes to cytosolic translocation of extracellular antigen for cross-presentation by dendritic cells. Proc Natl Acad Sci U S A 108: 16363-16368. [Crossref]
- Blachere NE, Darnell RB, Albert ML (2005) Apoptotic cells deliver processed antigen to dendritic cells for cross-presentation. PLoS Biol 3: e185. [Crossref]
- Shen L, Rock KL (2004) Cellular protein is the source of cross-priming antigen in vivo. Proc Natl Acad Sci U S A 101: 3035-3040. [Crossref]
- Murshid A, Gong J, Calderwood SK (2010) Heat shock protein 90 mediates efficient antigen cross presentation through the scavenger receptor expressed by endothelial cells-I. J Immunol 185: 2903-2917. [Crossref]
- Oura J, Tamura Y, Kamiguchi K, Kutomi G, Sahara H, et al. (2011) Extracellular heat shock protein 90 plays a role in translocating chaperoned antigen from endosome to proteasome for generating antigenic peptide to be cross-presented by dendritic cells. Int Immunol 23: 223-237. [Crossref]
- Tanaka T, Okuya K, Kutomi G, Takaya A, Kajiwara T, et al. (2015) Heat shock protein 90 targets a chaperoned peptide to the static early endosome for efficient cross-presentation by human dendritic cells. Cancer Sci 106: 18-24. [Crossref]
- Murshid A, Gong J, Calderwood SK (2014) Hsp90-peptide complexes stimulate antigen presentation through the class II pathway after binding scavenger receptor SREC-I. Immunobiology 219: 924-931. [Crossref]
- Krieg AM1 (2002) CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol 20: 709-760. [Crossref]
- Okuya K, Tamura Y, Saito K, Kutomi G, Torigoe T, et al. (2010) Spatiotemporal regulation of heat shock protein 90-chaperoned self-DNA and CpG-oligodeoxynucleotide for type I IFN induction via targeting to static early endosome. J Immunol 184: 7092-7099. [Crossref]
- Guiducci C, Ott G, Chan JH, Damon E, Calacsan C, et al. (2006) Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J Exp Med 203: 1999-2008. [Crossref]
- Shortman K, Liu YJ (2002) Mouse and human dendritic cell subtypes. Nat Rev Immunol 2: 151-161. [Crossref]
- van Eden W, van der Zee R, Prakken B (2005) Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol 5: 318-330. [Crossref]
- Shimada A, Kasatani T, Takei I, Maruyama T, Nomaguchi H, et al. (1996) Immune response to heat-shock protein correlates with induction of insulitis in I-E alpha d transgenic NOD mice. Diabetes 45: 165-169. [Crossref]
- van Eden W, Hauet-Broere F, Berlo S, Paul L, van der Zee R, et al. (2005) Stress proteins as inducers and targets of regulatory T cells in arthritis. Int Rev Immunol 24: 181-197. [Crossref]
- Beech JT, Siew LK, Ghoraishian M, Stasiuk LM, Elson CJ, et al. (1997) CD4+ Th2 cells specific for mycobacterial 65-kilodalton heat shock protein protect against pristane-induced arthritis. J Immunol 159: 3692-3697. [Crossref]
- Mattei D, Ozaki LS, de Silva LP (1988) A Plasmodium falciparum gene encoding a heat shock-like antigen related to the rat 78 Kd glucose-regulated protein. Nucleic Acids Res 16: 5204. [Crossref]
- Li Z, Menoret A, Srivastava P (2002) Roles of heat-shock proteins in antigen presentation and cross-presentation. Curr Opin Immunol 14: 45-51. [Crossref]
- Ishii T, Udono H, Yamano T, Ohta H, Uenaka A, et al. (1999) Isolation of MHC class I-restricted tumor antigen peptide and its precursors associated with heat shock proteins hsp70, hsp90, and gp96. J Immunol 162: 1303-1309. [Crossref]
- Basu S, Binder RJ, Ramalingam T, Srivastava PK (2001) CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 14: 303-313. [Crossref]
- Srivastava PK, DeLeo AB, Old LJ (1986) Tumor rejection antigens of chemically induced sarcomas of inbred mice. Proc Natl Acad Sci U S A 83: 3407-3411. [Crossref]
- Brás A, Aguas AP (1996) Diabetes-prone NOD mice are resistant to Mycobacterium avium and the infection prevents autoimmune disease. Immunology 89: 20-25. [Crossref]
- Birk OS, Douek DC, Elias D, Takacs K, Dewchand H, et al. (1996) A role of Hsp60 in autoimmune diabetes: analysis in a transgenic model. Proc Natl Acad Sci U S A 93: 1032-1037. [Crossref]
- Mackern-Oberti JP, Llanos C, Riedel CA, et al. (2015) Contribution of dendritic cells to the autoimmune pathology of systemic lupus erythematosus. Immunology 146: 497-507. [Crossref]
- Rekvig OP (2015) The anti-DNA antibody: origin and impact, dogmas and controversies. Nat Rev Rheumatol 11: 530-540. [Crossref]
- Minota S, Koyasu S, Yahara I, Winfield J (1988) Autoantibodies to the heat-shock protein hsp90 in systemic lupus erythematosus. J Clin Invest 81: 106-109. [Crossref]
- Ripley BJ, Isenberg DA, Latchman DS (2001) Elevated levels of the 90 kDa heat shock protein (hsp90) in SLE correlate with levels of IL-6 and autoantibodies to hsp90. J Autoimmun 17: 341-346. [Crossref]
- Saito K, Kukita K, Kutomi G, et al. (2015) Heat shock protein 90 associates with Toll-like receptors 7/9 and mediates self-nucleic acid recognition in SLE. Eur J Immunol 45: 2028-2041. [Crossref]
- Stephanou A, Latchman DS, Isenberg DA (1998) The regulation of heat shock proteins and their role in systemic lupus erythematosus. Semin Arthritis Rheum 28: 155-162. [Crossref]
- Liu Y, Ye J, Shin Ogawa L, Inoue T, Huang Q, et al. (2015) The HSP90 Inhibitor Ganetespib Alleviates Disease Progression and Augments Intermittent Cyclophosphamide Therapy in the MRL/lpr Mouse Model of Systemic Lupus Erythematosus. PLoS One 10: e0127361. [Crossref]
- Proia DA, Foley KP, Korbut T, Sang J, Smith D, et al. (2011) Multifaceted intervention by the Hsp90 inhibitor ganetespib (STA-9090) in cancer cells with activated JAK/STAT signaling. PLoS One 6: e18552. [Crossref]
- Guzhova I, Kislyakova K, Moskaliova O, Fridlanskaya I, Tytell M, et al. (2001) In vitro studies show that Hsp70 can be released by glia and that exogenous Hsp70 can enhance neuronal stress tolerance. Brain Res 914: 66-73. [Crossref]
- Tonkiss J, Calderwood SK (2005) Regulation of heat shock gene transcription in neuronal cells. Int J Hyperthermia 21: 433-444. [Crossref]
- Chen S, Brown IR (2007) Translocation of constitutively expressed heat shock protein Hsc70 to synapse-enriched areas of the cerebral cortex after hyperthermic stress. J Neurosci Res 85: 402-409. [Crossref]
- Calderwood SK (2005) Evolving connections between molecular chaperones and neuronal function. Int J Hyperthermia 21: 375-378. [Crossref]
- Bechmann I, Galea I, Perry VH (2007) What is the blood-brain barrier (not)? Trends Immunol 28: 5-11. [Crossref]
- Ransohoff RM, Engelhardt B (2012) The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol 12: 623-635. [Crossref]
- Yshii L, Gebauer C, Bernard-Valnet R, Liblau R (2015) Neurons and T cells: Understanding this interaction for inflammatory neurological diseases. Eur J Immunol 45: 2712-2720. [Crossref]
- Mahad DH, Trapp BD, Lassmann H (2015) Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 14: 183-193. [Crossref]
- Cwiklinska H, Mycko MP, Luvsannorov O, Walkowiak B, Brosnan CF, et al. (2003) Heat shock protein 70 associations with myelin basic protein and proteolipid protein in multiple sclerosis brains. Int Immunol 15: 241-249. [Crossref]
- Mansilla MJ, Montalban X, Espejo C (2012) Heat shock protein 70: roles in multiple sclerosis. Mol Med 18: 1018-1028. [Crossref]
- D'Souza SD, Antel JP, Freedman MS (1994) Cytokine induction of heat shock protein expression in human oligodendrocytes: an interleukin-1-mediated mechanism. J Neuroimmunol 50: 17-24. [Crossref]
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, et al. (2000) Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2: 469-475. [Crossref]
- Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES (2000) Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol 2: 476-483. [Crossref]
- Saibil H (2013) Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol 14: 630-642. [Crossref]
- Pratt WB, Gestwicki JE, Osawa Y, Lieberman AP (2015) Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu Rev Pharmacol Toxicol 55: 353-371. [Crossref]
- Rajdev S, Hara K, Kokubo Y, Mestril R, Dillmann W, et al. (2000) Mice overexpressing rat heat shock protein 70 are protected against cerebral infarction. Ann Neurol 47: 782-791. [Crossref]
- Stadelmann C, Ludwin S, Tabira T, Guseo A, Lucchinetti CF, et al. (2005) Tissue preconditioning may explain concentric lesions in Baló's type of multiple sclerosis. Brain 128: 979-987. [Crossref]
- Lehner T, Bergmeier LA, Wang Y, Tao L, Sing M, et al. (2000) Heat shock proteins generate beta-chemokines which function as innate adjuvants enhancing adaptive immunity. Eur J Immunol 30: 594-603. [Crossref]
- Panjwani NN, Popova L, Srivastava PK (2002) Heat shock proteins gp96 and hsp70 activate the release of nitric oxide by APCs. J Immunol 168: 2997-3003. [Crossref]
- Kuppner MC, Gastpar R, Gelwer S, Nössner E, Ochmann O, et al. (2001) The role of heat shock protein (hsp70) in dendritic cell maturation: hsp70 induces the maturation of immature dendritic cells but reduces DC differentiation from monocyte precursors. Eur J Immunol 31: 1602-1609. [Crossref]
- Fleshner M, Johnson JD (2005) Endogenous extra-cellular heat shock protein 72: releasing signal(s) and function. Int J Hyperthermia 21: 457-471. [Crossref]
- Tezel G, Edward DP, Wax MB (1999) Serum autoantibodies to optic nerve head glycosaminoglycans in patients with glaucoma. Arch Ophthalmol 117: 917-924. [Crossref]
- Kremmer S, Kreuzfelder E, Klein R, Bontke N, Henneberg-Quester KB, et al. (2001) Antiphosphatidylserine antibodies are elevated in normal tension glaucoma. Clin Exp Immunol 125: 211-215. [Crossref]
- Yang J, Patil RV, Yu H, Gordon M, Wax MB (2001) T cell subsets and sIL-2R/IL-2 levels in patients with glaucoma. Am J Ophthalmol 131: 421-426. [Crossref]
- Wax MB, Tezel G, Yang J, Peng G, Patil RV, et al. (2008) Induced autoimmunity to heat shock proteins elicits glaucomatous loss of retinal ganglion cell neurons via activated T-cell-derived fas-ligand. J Neurosci 28: 12085-12096. [Crossref]