Regucalcin was initially discovered in 1978 as a unique calcium-binding protein that is a suppressor in intracellular calcium signaling. The regucalcin gene (rgn) is localized on the X chromosome and is identified in over 15 species consisting of regucalcin family. Regucalcin has been demonstrated to play a multifunctional role in cell regulation of calcium homeostasis, signal transduction, nuclear gene expression, cell proliferation and apoptosis in various types of cells and tissues. Moreover, regucalcin plays a pathophysiologic role in hyperlipidemia and diabetes. Liver regucalcin gene expression is stimulated through action of insulin in liver cells and decreased in type I diabetic model animals. Overexpreesion of regucalcin reveals hepatic insulin resistance, decreased liver triglyceride, total cholesterol and glycogen contents in the liver of rats, inducing a hyperlipidemia. Liver leptin and adiponectin mRNA expressions are decreased by overexpression of regucalcin. Deficiency of regucalcin induces an impairment of glucose tolerance and liver lipid accumulation in mice, and it is associated with the development and progression of nonalcoholic fatty liver disease and fibrosis in human patients. Regucalcin may be a key molecule in lipid metabolic disorder implicated in obesity and diabetes.
Regucalcin, Adipogenesis, Insulin resistance, Lipid metabolic disorder, Obesity, Diabetes
Obesity and diabetes are currently a major health problem worldwide with growing in prevalence. The incidence of metabolic disease, including type 2 diabetes with obesity, is increased to epidemic levels. Obesity and diabetes induce secondary diseases with various pathophysiologic states, which are important in clinical aspects including cardiovascular disease, neural disturbance, kidney disease, osteoporosis and cancer [1-6].
Obesity is based on stimulation of adipogenesis. Bone marrow mesenchymal stem cells are multipotent cells, which among other cell lineages, and give to differentiate into adipocytes, osteoblasts, chondrocytes and myoblasts [1]. This occurs through cross talk between complex signaling pathways including those derived from bone morphogenic proteins, winglesstype MMTV integration site (Wnt) proteins, hedgehogs, delta/jagged proteins, fibroblastic growth factors, insulin, insulin-like growth factors, and transcriptional regulators of adipocyte and osteoblast differentiation including peroxisome proliferators-activated receptor-gamma (PPARγ) and runt-related transcription factor 2 (Runx2) [1-3]. Insulin, which is secreted by feeding, stimulates adipogenesis from bone marrow mesenchymal stem cells. In addition, bone marrow adiposity and mature adipocytes with obesity greatly produces tumor necrosis factor-α (TNF-α), an inflammatory cytokine [4]. This TNF-α may cause insulin resistance that leads to type 2 diabetes.
Osteoporosis, which bone mass is dramatically reduced after menopause, has also been shown to induce with obesity, diabetes (type I and II) and inflammatory disease [7,8]. Type 1 and type 2 diabetes have been associated with increased fracture risk. Osteoporosis and obesity are now thought to be closely related and to share several features. Osteoporosis and obesity are now thought to be closely related and to share several features [1,2,4]. One of shared features for obesity with osteoporosis is that osteoblasts and adipocytes differentiate from a common precursor cell in the bone marrow mesenchymal stem cells. There is an inverse relationship between differentiation of mesenchymal stem cells to osteoblasts and adipocytes. Secondary causes of osteoporosis including obesity and diabetes are associated with bone marrow adiposity, which greatly produces tumor necrosis factor-α (TNF-α), an inflammatory cytokine [4,5].
Various hormones and cytokines, which include leptin, adiponectin, insulin, epinephrine, cortisol, glucagon, TNF-α and other factors, are well known as key molecules that relate to obesity and diabetes. Disturbance of these factors may play an important role in pathophysiologic conditions of obesity and diabetes. Moreover, regucalcin, which is a suppressor protein of intracellular signaling systems [9], has been proposed to be a key molecule in obesity and diabetes associated with osteoporosis [10-12]. In addition, regucalcin has been demonstrated to stimulate adipogenesis in mouse bone marrow cell culture in vitro [12], suggesting an involvement as a stimulatory factor in adipogenesis. This review will discuss an involvement of regucalcin as a novel protein in obesity and diabetes.
Regucalcin, which was discovered in 1978 [13,14], plays a multifunctional role as a suppressor protein in signal transduction in various cell types and plays a cell physiologic role in maintaining cell homeostasis for various stimuli [9,15-17]. Cytoplasmic regucalcin localizes into the nucleus, and it suppresses nuclear protein kinase and protein phosphatase activities and deoxyribonucleic acid and ribonucleic acid synthesis and regulates gene expression for various proteins [15]. Regucalcin has also been shown to suppress protein synthesis and activate proteolysis, suggesting a role as suppressor in protein turnover [9]. Moreover, overexpression of endogenous regucalcin has been demonstrated to suppress cell proliferation and apoptosis induced through multisignaling pathways in various cell types [16,17]. Regucalcin has been demonstrated to play a pivotal role in cell regulation in various type cell and tissues including liver, kidney, heart, brain and bone [9, 18-20].
Yamaguchi et al. generated regucalcin transgenic rats that reveal overexpression of endogenous regucalcin, and this animal was found to induce hyperlipidemia associated with osteoporosis [21]. Exogenous regucalcin has been shown to stimulate osteoclastic bone resorption and suppress osteoblastic bone formation, thereby decreasing bone mass [19]. Moreover, exogenous regucalcin was found to suppress osteoblastogenesis and stimulate adipogenesis in mouse bone marrow culture in vitro [11]. Regucalcin may stimulate differentiation from bone marrow mesenchymal stem cells to adipocytes, supporting the view that regucalcin plays a regulatory role in adipogenesis.
Regucalcin transgenic (TG) rats, which overexpresses endogenous regucalcin, have been shown to induce a remarkable of bone loss associated with increase in serum triglyceride and high-density lipoprotein (HDL)-cholesterol concentrations at the age of 36 weeks in vivo [22,23]. Serum free fatty acid, triglyceride, cholesterol or HDL-cholesterol concentrations were markedly increased in regucalcin TG male and female rats at 14-50 weeks of age [23]. Thus, hyperlipidemia associated with bone loss was found to induce in regucalcin TG rats with increasing age. This animal may be a useful tool in the aspects of lipid metabolic disorder and osteoporosis.
The change in lipid components in the adipose and liver tissues of regucalcin TG rats with increasing age has also been shown in vivo [24]. Regucalcin was expressed in the adipose tissues of normal rats [24]. Triglyceride content in the adipose tissues was increased in regucalcin TG rats with aging [24]. This may be an implication with adipogenesis that is enhanced by regucalcin [11]. Liver triglyceride, total cholesterol, free fatty acid and glycogen contents were decreased in regucalcin TG rats [24]. The expression of regucalcin in the liver tissues was enhanced in regucalcin TG rats [24]. Regucalcin suppressed the activations of glycogen particulate phosphorylase a, cytoplasmic pyruvate kinase, and fructose 1,6-diphosphatase in rat liver [9]. Regucalcin may suppress glycogen synthesis in the liver and stimulate glycogenolysis in regucalcin TG rats. As the result, lipid synthesis may be stimulated in the liver tissues of the TG rats in vivo.
Leptin and adiponectine are adipokines that are involved in lipid metabolism [25]. Leptin mRNA expression in the adipose or liver tissues was found to decrease in regucalcin TG rats with aging [24]. Adiponectin mRNA expression was not changed in the adipose tissues of the TG rats, while its level was decreased in the liver tissues [24]. These decreases may be partly involved in hyperlipidemia induced in regucalcin TG rats. Regucalcin may play an important role in the disorder of lipid metabolism in the liver. The role of regucalcin in the regulation of lipid metabolism and implication with obesity and diabetes is summarized in Figure 1.
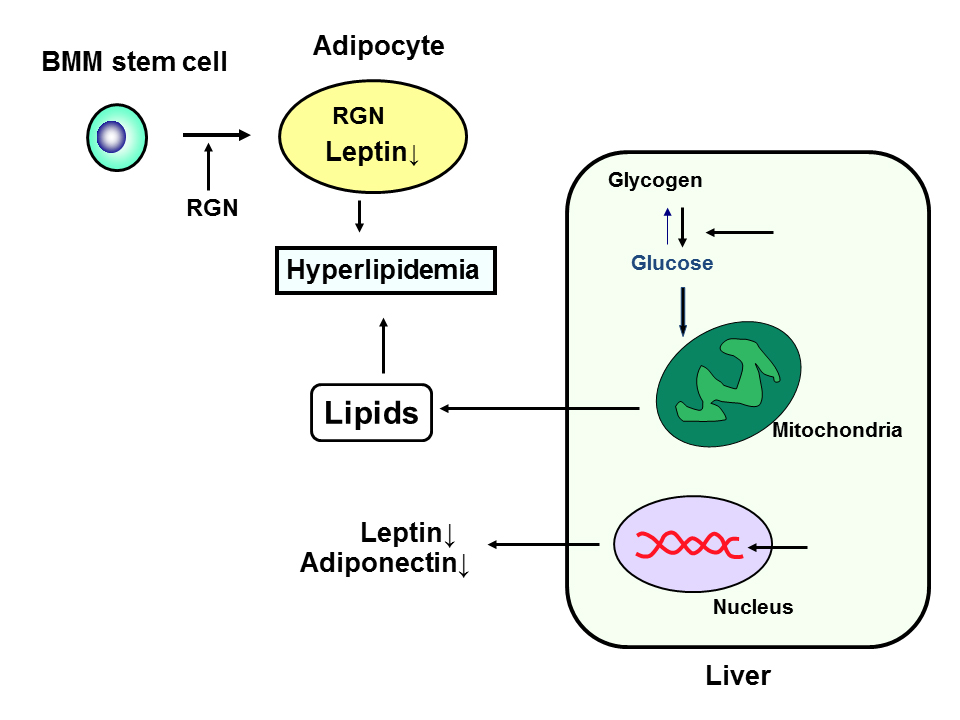
Figure 1. Role of regucalcin (RGN) in the regulation of lipid metabolism and implication with obesity and diabetes. RGN stimulates differentiation from bone marrow mesenchymal (BMM) stem cells to adipocytes, and it enhances adipogenesis and enhances triglyceride in the adipocytes. RGN suppresses leptin expression in adipocytes that regulate feeding. RGN stimulates glycolysis and production of in the liver cells, and it suppress adiponectin and leptin mRNA expression in the liver cells.
Hyperlipidemia has been reported to induce in various animal models; lipoprotein lipase- deficient mice [26], low-density lipoprotein (LDL) receptor-deficient mice [27], apolipoprotein C3-KO mice [28], apolipoprotein C1 TG mice [29], very LDL lipoprotein receptor KO mice [30], cholesterol 7 alpha-hydroxylase-deficient mice [31], apoE-deficient mice [32] and hepatic myr-Akt overexpressing mice [33]. These animal models for hyperlipidemia are based on molecules that are regulated to lipid metabolism. Regucalcin is a novel protein molecule that regulates lipid metabolism [12].
Regucalcin plays an important role in the regulation of glucose and lipid metabolism [12]. Fasting induced a decrease in regucalcin mRNA expression in rat liver in vivo, and this decrease was restored after re-feeding in rats in vivo [34], suggesting that feeding is a physiologic factor in the regulation of the regucalcin gene expression. In addition, oral administration of glucose to fasted rats caused a significant increase in hepatic regucalcin mRNA expression [34], suggesting an involvement of insulin secreted from pancreatic cells after glucose administration. Moreover, hepatic regucalcin mRNA expression was clearly elevated after a single subcutaneous administration of insulin to fasted rats in vivo [34]. In fact, insulin was demonstrated to directly stimulate regucalcin mRNA and protein expressions in human hepatoma cells (HepG2) in vitro [35]. Insulin, which is related to metabolism of blood glucose after feeding, stimulated regucalcin gene expression in liver cells. Hepatic regucalcin expression was markedly decreased after a single subcutaneous administration of streptozotocin that induces type 1 diabetes [36]. These findings may support the view that regucalcin gene expression is enhanced by insulin, and that regucalcin may be involved in liver metabolic disorder related to diabetes.
Deficiency of regucalcin has been reported to cause an impairment of glucose tolerance in regucalcin knockout (KO) mice [37,38]. Regucalcin KO mice caused a significant increase in blood glucose concentration and a decrease in serum insulin levels after glucose administration compared with wild-type mice in vivo [38], suggesting that regucalcin participates in the regulation of glucose metabolism related to insulin action. Regucalcin deficiency in mice caused an accumulation of neutral lipids and phospholipids in the liver and shortens the life span [37]. Hepatocytes, which were obtained from regucalcin KO mice at 12 months of age, was shown to contain many lipid droplets, abnormally enlarged mitochondria with indistinct cristae, and enlarged lysosomes filled with electron-dense bodies in the electron microscope as compared with that of wild-type mice [37]. Hepatic neutral lipids, total phospholipids, total triglyceride and cholesterol in regucalcin KO mice were found to markedly increase than those from age-matched wild-type mice [37].
Regucalcin was not expressed in hepatic stellate cells (HSCs) of both wild type and regucalcin KO mice [39,40]. Peroxisome proliferator-activated receptor-gamma (PPAR-γ) was up-regulated in the liver of regucalcin KO mice [39]. Numerous HSCs was hypertrophic and contained abundant microvesicular lipid droplets in the liver cytoplasm of aged regucalcin KO mice [39,40]. The expression of PPAR-γ, which is a protein related to lipid metabolism and HSC quiescence, was also found to increase in hypertrophic HSCs of regucalcin KO mice [39]. Deficiency of regucalcin leads to accumulation of liver lipid components.
Insulin resistance may be modeled in culture system by using cloned rat hepatoma H4-II-E cells cultured with insulin and TNF-α in vitro [41]. This in vitro model nicely mimicsed insulin resistance in human type 2 diabetic mellitus. When H4-II-E cells were cultured in the presence of TNF-α plus insulin in vitro, regucalcin was identified as an important protein, which is involved in insulin resistance, by proteome analysis [41]. Regucalcin may be a key molecule that is related to insulin resistance.
Regucalcin, moreover, has been demonstrated to stimulate glucose utilization and lipid production in H4-II-E cells in vitro [42]. Overexpression of endogenous regucalcin was found to stimulate the production of triglyceride and free fatty acid in H4-II-E cells cultured with or without the supplementation of glucose in the absence of insulin [42]. Regucalcin may stimulate lipid production that is linked to glucose metabolism in liver cells in vitro. The effect of insulin, which enhances medium glucose consumption, triglyceride and free fatty acid productions in liver cells cultured with glucose supplementation, was suppressed by overexpression of regucalcin in vitro [43].
Insulin resistance in the liver is associated with the pathogenesis of nonalcoholic fatty liver disease (NAFLD). Change in hepatic regucalcin levels may be associated with the development and progression of NAFLD [44]. Patients with NAFLD had a significant lower level of hepatic regucalcin [44]. Hepatic regucalcin levels decreased in a fibrosis stage-dependent manner and were correlated negatively with the homeostasis model assessment of insulin resistance, the net electronegative charge modified-LDL, and type IV collagen 7S [44]. Both serum large very LDL and very small LDL levels were elevated in patients with NAFLD [44]. Whether or not the decrease in hepatic regucalcin in human patients is a result or a cause of cirrhosis remains to be elucidated, however [44]. Insulin resistance in the liver is associated with the pathogenesis of NFLD, suggesting an involvement in lipid metabolic disorder.
Molecular mechanism by which regucalcin regulates glucose metabolism related insulin action has been elucidated. Overexpression of regucalcin did not reveal stimulatory effects on the gene expression of enzymes, which are related to glucose and lipid metabolism, including acetyl-CoA carboxylase, HMG-CoA reductase, glucokinase and pyruvate kinase in liver cells after culture with or without glucose supplementation in the presence of insulin [42]. Overexpression of regucalcin increased the expression of glucose transporter 2 (GLUT 2) mRNA to enhance glucose utilization in the liver cells [42,43], and it was found to suppress the expression of insulin receptor (Insr) or phosphatidylinositol 3-kinase (PI3K) mRNAs that are an insulin signaling-related protein [42,43]. Suppressive effects of regucalcin on the expression of Insr and PI3K mRNAs may play an important role in insulin resistance in liver cells overexpressing endogenous regucalcin. Moreover, regucalcin may suppress signal transduction pathways related to insulin action in liver cells.
Regucalcin may play a physiological role in lipid and glucose metabolism in the adipocytes and liver those are implicated in obesity and diabetes. Regucalcin, which its gene expression is enhanced by insulin, is identified as a molecule related to insulin resistance in liver cells. Deficiency of regucalcin impairs glucose tolerance and induces liver lipid accumulation. Overexpression of regucalcin stimulates hepatic glycolysis and lipid production. Disturbance of hepatic regucalcin gene expression may leads to disorders of lipid metabolism and insulin resistance in liver tissues, inducing a hyperlipidemia. Regucalcin may play a pathophysiologic role as a regulatory protein implicated in obesity and diabetes. Regucalcin may be a target molecule for therapy of these diseases.
The author has no conflicts of interest.
The author was supported by the Foundation for Biomedical Research on Regucalcin.
- Minguel JJ, Erices A, Conget P (2001) Mesenchymal stem cells. Exp Biol Med 226: 507-520.
- Muruganandan S, Roman AA, Sinal CJ (2009) Adipocyte differentiation of bone marrow-derived mesenchymal stem cells: cross talk with the osteoblastogenesis program. Cell Mol Life Sci 66: 236-253.
- Laudes M (2011) Role of WNT signaling in the determination of human mesenchymal stem cells into preadipocytes. J Mol Endocrinol 46: R65-72.
- Gharibi B, Abraham AA, Ham J, Evans BA (2011) Adenosine receptor subtype expression and activation influence the differentiation of mesenchymal stem cells to osteoblasts and adipocytes. J Bone Miner Res 26: 2112-2124.
- Kawai M, Rosen CJ (2010) PPARy: a circadian transcription factor in adipogenesis and osteogenesis. Nat Rev Endocrinol 6: 629-636.
- Rosen CJ, Bouxsein ML (2006) Mechanisms of disease: its osteoporosis the obesity of bone? Nat Clin Pract Rheumatol 2: 35-43.
- Leslie WD, Rubin MR, Schwartz AV, Kanis JA (2012) Type 2 diabetes and bone. J Bone Miner Res 27: 2231-2237.
- Nielson CM, Srikanth P, Orwoll ES (2012) Obesity and fracture in men and women: An epidemiologicperspective. J Bone Miner Res 27: 1-10.
- Yamaguchi M (2011) Regucalcin and cell regulation: role as a suppressor protein in signal transduction. Mol Cell Biochem 353: 101-137.
- Yamaguchi M (2010) Regucalcin and metabolic disorders: osteoporosis and hyperlipidemia are induced in regucalcin transgenic rats. Mol Cell Biochem 341: 119-133.
- Yamaguchi M, Weitzmann MN, Baile CA, Murata T (2012) Exogenous regucalcin suppresses osteoblastogenesis and stimulates adipogenesis in mouse bone marrow culture. Integr Biol (Camb) 4: 1215-1222.
- Yamaguchi M, Murata T (2013) Involvement of regucalcin in lipid metabolism and diabetes. Metabolism 62:1045-1051.
- 13. Yamaguchi M, Yamamoto T (1978) Purification of calcium binding substance from soluble fraction of normal rat liver. Chem Pharm Bull (Tokyo) 26: 1915- 1918.
- Yamaguchi M, Sakurai T (1991) Inhibitory effect of calcium-binding protein regucalcin on Ca2+-activated DNA ragmentation in rat liver nuclei. FEBS Lett 279: 281-284 .
- Yamaguchi M (2013) Role of regucalcin in cell nuclear regulation: Involvement as a transcription factor. Cell Tissue Res 354:331-341.
- Yamaguchi M (2013) Suppressive role of regucalcin in liver cell proliferation: Involvement in carcinogenesis. Cell Prolif 46: 243-253.
- Yamaguchi M (2013) The anti-apoptotic effect of regucalcin is mediated through multisignaling pathways. Apoptosis 18: 1145–1153.
- Yamaguchi M (2012) Role of regucalcin in brain calcium signaling: Involevement with aging. Integr Biol (Cambr) 4: 825-837.
- Yamaguchi M (2014) Role of regucalcin in bone homeostasis: Involvement as a novel cytokine. Integr Biol (Cambr) 6: 258-266.
- Yamaguchi M (2014) Re2021 Copyright OAT. All rights reservcalcium signaling: Insight into cardiac failaure (Review). Biomed Reports 2:303-308.
- Yamaguchi M, Morooka Y, Misawa H, Tsurusaki Y, Nakajima R (2002) Role of endogenous regucalcin in transgenic rats: Suppression of kidney cortex cytosolic protein phosphatase activity and enhancement of heart muscle microsomal Ca2+-ATPase activity. J Cell Biochem 86: 520-529.
- Yamaguchi M, Misawa H, Uchiyama S, Morooka Y, Tsurusaki Y (2002) Role of endogenous regucalcin in bone metabolism: bone loss in induced in regucalcin transgenic rats. Int J Mol Med 10: 377-383.
- Yamaguchi M, Igarashi A, Uchiyama S, Sawada N (2004) Hyperlipidemia is induced in regucalcin transgenic rats with increasing age. Int J Mol Med 14: 647-651.
- Yamaguchi M, Nakagawa T (2007) Change in lipid components in the adipose and liver tissues of regucalcin transgenic rats with increasing age: suppression of leptin and adiponectin gene expression. Int J Mol Med 20: 323-328.
- Ronti T, Lupattelli G, Mannarino E (2006) The endocrine function of adipose tissue: an update. Clin Endocrinol (Oxf) 64: 355-365.
- Weinstock PH, Bisgaier CL, Aalto-Setälä K, Radner H, Ramakrishnan R, et al. (1995) Severe hypertriglyceridemia, reduced high density lipoprotein, and neonatal death in lipoprotein lipase knockout mice. Mild hypertriglyceridemia with impaired very low density lipoprotein clearance in heterozygotes. J Clin Invest 96: 2555-2568.
- Lichtman AH, Clinton SK, Iiyama K, Connelly PW, Libby P, et al. (1999) Hyperlipidemia and atherosclerotic lesion development in LDL receptor-deficient mice fed defined semipurified diets with and without cholate. Arterioscler Thromb Vasc Biol 19: 1938-1944.
- Jong MC, Havekes LM (2000) Insights into apolipoprotein C metabolism from transgenic and gene-targeted mice. Int J Tissue React 22: 59-66.
- Koopmans SJ, Jong MC, Que I, Dahlmans VE, Pijl H, et al. (2001) Hyperlipidaemia is associated with increased insulin-mediated glucose metabolism, reduced fatty acid metabolism and normal blood pressure in transgenic mice overexpressing human apolipoprotein C1. Diabetologia 44: 437-443.
- Yagyu H, Lutz EP, Kako Y, Marks S, Hu Y, et al. (2002) Very low density lipoprotein (VLDL) receptor-deficient mice have reduced lipoprotein lipase activity. Possible causes of hypertriglyceridemia and reduced body mass with VLDL receptor deficiency. J Biol Chem 277: 10037-10043.
- Chen JY, Levy-Wilson B, Goodart S, Cooper AD (2002) Mice expressing the human CYP7A1 gene in the mouse CYP7A1 knock-out background lock induction of CYP7A1 expression by cholesterol feeding and have increased hypercholesterolemia when fed a high fat diet. J Biol Chem 277: 42588-42595.
- Fazio S, Linton MF (2001) Mouse models of hyperlipidemia and atherosclerosis. Front Biosci 6: D515-525.
- Ono H, Shimano H, Katagiri H, Yahagi N, Sakoda H, et al. (2003) Hepatic Akt activation induces marked hypoglycemia, hepatomegaly, and hypertriglyceridemia with sterol regulatory element binding protein involvement. Diabetes 52: 2905-2913.
- Yamaguchi M, Oishi K, Isogai M (1995) Expression of hepatic calcium-binding protein regucalcin mRNA is elevated by refeeding of fasted rats: involvement of glucose, insulin and calcium as stimulating factors. Mol Cell Biochem 142: 35-41.
- Murata T, Shinya N, Yamaguchi M (1997) Expression of calcium-binding protein regucalcin mRNA in the cloned human hepatoma cells (HepG2): stimulation by insulin. Mol Cell Biochem 175: 163-168.
- Isogai M, Kurota H, Yamaguchi M (1997) Hepatic calcium-bindingbprotein regucalcin concentration is decreased by streptozotocin-diabetic state and ethanol ingestion in rats. Mol Cell Biochem 168: 67-72.
- Ishigami A, Kondo Y, Nanba R, Ohsawa T, Handa S, et al. (2004) SMP30 deficiency in mice causes an accumulation of neutral lipids and phospholipids in the liver and shortens the life span. Biochem Biophys Res Commun 315: 575-580.
- Hasegawa G, Yamasaki M, Kadono M, Tanaka M, Asano M, et al. (2010) Senescence marker protein-30/gluconolactonase deletion worsens glucose tolerance through impairment of acute insulin secretion. Endocrinology 151: 529-536.
- Park JK, Ki MR, Lee HR, Hong IH, Ji AR, et al. (2010) Vitamin C deficiency attenuates liver fibrosis by way of up-regulated peroxisome proliferator-activated receptor-gamma expression in senescence marker protein 30 knockout mice. Hepatology 51:1766-1777.
- Hong IH, Han JY, Goo MJ, Hwa SY, Ki MR, et al. (2012) Ascorbic acid deficiency accelerates aging of hepatic stellate cells with up-regulation of PPARγ. Histol Histopathol 27:171-179.
- Solomon SS, Buss N, Shull J, Monnier S, Majumdar G, et al. (2005) Proteome of H-411E (liver) cells exposed to insulin and tumor necrosis factor-alpha: analysis of proteins involved in insulin resistance. J Lab Clin Med 145: 275-283.
- Nakashima C, Yamaguchi M (2006) Overexpression of regucalcin enhances glucose utilization and lipid production in cloned rat hepatoma H4-II-E cells: Involvement of insulin resistance. J Cell Biochem 99: 1582-1592.
- Nakashima C, Yamaguchi M (2007) Overexpression of regucalcin suppresses gene expression of insulin signaling-related proteins in cloned rat hepatoma H4-II-E cells: involvement of insulin resistance. Int J Mol Med 20: 709-716.
- Park H, Ishigami A, Shima T, Mizuno M, Maruyama N, et al. (2010) Hepatic senescence marker protein-30 is involved in the progression of nonalcoholic fatty liver disease. J Gastroenterol 45: 426-434.