Abstract
The complete acronymous of MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) is presented only in about 10% of the cases. Stroke-like episodes of MELAS syndrome, often occur before 40 years of age. Clinical manifestations vary from asymptomatic to progressive muscle weakness; with lactic acidosis, cognitive dysfunction, seizures, stroke-like episodes, mitochondrial encephalopathy and premature death. We present a case of an 18-year-old male with a history of parkinsonism movements during the last year. In the emergency room, he presented generalized weakness, global aphasia, and parkinsonism movements. Persistence of clinical manifestations over a three month period was observed because of lack of financial support. Physical exam revealed global aphasia, upper limbs spasticity with anteflexion of the right hand, generalized muscle atrophy; spontaneous myoclonus of variable duration, alternating with ataxia, dystonia, parkinsonism movements, choreoathetosis and spastic paraplegia. CT brain scan showed a hypodense lesion in the right putamen. Brain MRI revealed an early infarction in the right putamen and mild cerebral edema. Autoimmune and infectious tests were normal. Serum levels of lactic acid: 7.7 (0.7-2.1). Given the high levels of lactic acid and the radiological findings, a mitochondrial DNA analysis was performed which detected a mutation in A3243G, confirming the diagnosis. Despite treatment with pyridoxine, Coenzyme Q-10, and L-arginine, the patient had a poor neurological response with a fatal outcome.
Key words
MELAS (myopathy, encephalopathy, lactic acidosis, stroke-like episodes), parkinsonism, ataxia, dystonia, choreoathetosis
Introduction
Mitochondrial encephalopathies are a group of multisystemic diseases characterized by a broad range of biochemical and genetic mitochondrial disorders with variable inheritance [1]. Stroke-like episodes that define the MELAS syndrome often occur before 40 years of age. The clinical manifestations are variable; from asymptomatic with normal development, to progressive muscle weakness with lactic acidosis, cognitive dysfunction, seizures, stroke-like episodes, mitochondrial encephalopathy and premature death [1]. The main causes of stroke-like episodes associated with MELAS are unknown. Two possible mechanisms are proposed: Mitochondrial angiopathy or mitochondrial cytopathy or both. Ischemic vascular mitochondrial mechanism caused by angiopathy sometimes improves endothelial dysfunction with L-arginine in MELAS syndrome [2]. Since the discovery of the mutation in mitochondrial DNA tRNALeu (UUR) of A3243G, the cytopathic mechanism of the defect of oxidative phosphorylation in the neuron, glial cells, and endothelial cells has been demonstrated. This gene is in the respiratory chain of MELAS syndrome described in 80% of the cases [2-4].
Case report
An 18-year-old male with a history of encephalitis of unknown etiology one year prior to admission; presented to the emergency room with generalized weakness and global aphasia. Due to lack of financial resources the patient persists with the same clinical manifestations for three months leaving sequels with parkinsonism type movements. Physical exam revealed global aphasia, spasticity of the upper limbs with anteflexion of the right hand, generalized muscle atrophy, spontaneous myoclonus of variable duration; alternating with ataxia, dystonia, choreoathetosis and spastic paraplegia. Laboratory tests: Glucose: 80 mg/dl, Hb: 11.57 g/dl, Ht: 32.80%, PLT: 180,500, DHL: 345 mg/dl, total CPK: 1267. Lumbar puncture with bacterial, viral and fungal results were normal. Anti-DNA, P- ANCA and C-ANCA, complement C3, complement C4, Anti RNP, anticardiolipin IgM and IgG antibodies; protein-C and protein-S, copper and ceruloplasmin were unremarkable. Serum lactic acid: 7.7 (0.7-2.1). CT brain scan: hypodense lesion in the right putamen (Figure 1). Brain MRI: a presence of an early infarction in the right putamen and mild cerebral edema (Figure 2). Electromyogram and funduscopy were also normal.
EEG: diffuse encephalopathy (Figure 3). Electrocardiogram: dilated right ventricle. Muscle biopsy: Gomori staining was negative. Given the high levels of lactic acid and the radiological findings, a mitochondrial DNA analysis was performed which detected a mutation in A3243G, confirming the diagnosis. Despite treatment with pyridoxine, Coenzyme Q-10, and L-arginine, the patient had a poor neurological response with a fatal outcome.
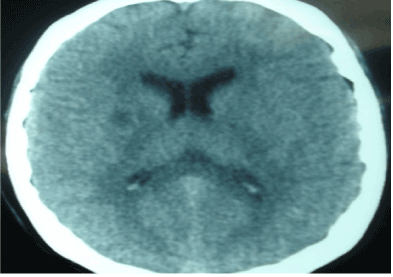
Figure 1. CT brain scan with with evidence of right hypodense lesion.
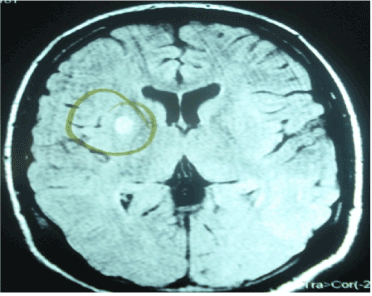
Figure 2. Brain MRI showed an earlier infarct area in the right putamen and mild cerebral edema.
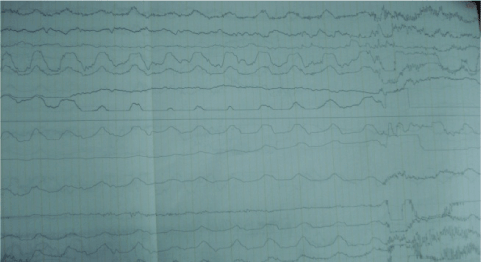
Figure 3. Electroencephalogram with diffuse encephalopathy.
Discussion
Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) is a maternally inherited multisystem disorder caused by mutations in mitochondrial DNA [5-8]. The main feature of this syndrome is the occurrence of stroke-like episodes that result in hemiparesis, hemianopsia, or cortical blindness. Plasma lactate levels> 2.5 mmol/L have a sensitivity and specificity of 62 and 100% for the diagnosis of a primary mitochondrial disease [9]. Neuropathy is a common involvement in mitochondrial diseases that presents in 37% of the cases [6]. Axonal neuropathy was the most common type (70%); motor forms predominated (50% pure motor, 30% sensorimotor and 20% pure sensitive), and greater severity is found in sensitive forms. Even though there are well-defined clinical syndromes, the main characteristic is the heterogeneity of the manifestations. This is partly conditioned by the heteroplasmic phenomena, mitotic segregation and threshold effect. Virtually any symptom or group of symptoms related to the involvement of any system, organ or tissue may reflect mitochondrial dysfunction. The following facts are particularly suggestive:
- Evidence of variable multisystemic progressive disorder of the central nervous system, peripheral nervous system, eyes, audition, heart and striated muscles.
- Progressive external ophthalmoplegia, especially if associated with retinitis pigmentosa.
- Association of myoclonus and ataxia.
- Cerebellar ataxia with proprioceptive sensory disturbances.
- Muscle weakness and exercise intolerance associated with a neurological syndrome.
- Partially recurrent and progressive neurological events (stroke-like), such as hemiparesis, hemianopsia, cortical blindness or a migraine.
- Short stature, Diabetes mellitus and progressive hearing loss.
A possible maternal inheritance can be suspected by the presence of "soft signs", such as short stature, deafness and migraines within family members of the maternal side. The workup usually required to establish the scope of the study process include fundus examination, EEG, auditory evoked potentials, somatosensory potentials, electroretinogram, visual evoked potentials, EMG and electro neuro graphic research. Neuroimaging tests particularly CT brain and, especially brain spectroscopic MRI can be very useful [10]. Stroke-like episodes in the posterior region of the brain and calcification of the base are often present in MELAS [10]. CT scan may show cerebral lesions of low attenuation in the occipital lobes. MRI may demonstrate T2-hyperintense lesions, predominantly in the cortex of the occipital and parietal lobes [11]. Staining with modified Gomori trichrome or better sensitivity for succinate dehydrogenase (SDH) may reveal the existence of the primary marker, the ragged red fibers (RRF). Its absence does not rule out mitochondrial disease, since this can depend on the developmental stage of the disease or the existence of a small proportion of DNA not involving mutated mitochondrial proliferation sufficient to form RRF. The results of the muscle biopsy in our patient with Gomori was normal. Determination of lactic acid and pyruvic blood predominates especially in CNS, CSF. A) one hour after ingestion of the standard breakfast (there was a significant increase of lactic acid greater than 5 mg/dL from baseline). MELAS syndrome is suspected in patients with frequent episodes of migraine, vomiting, fatigue or refractory epilepsy, especially when there is a maternal family history of diabetes, deafness, epilepsy, and dementia. They may also present with proteinuria in non nephrotic range, and the most characteristic histopathologic finding is a focal segmental glomerulosclerosis [12]. In our case, the molecular genetic analysis showed an A3243G mutation in the heteroplasmic mtDNA gene tRNALeu previously described [3]. The percentage of mutated molecules detected is relatively small (23-50%) (3). Currently, at least eight known mtDNA mutations are associated with MELAS syndrome, but the most prevalent in 80-85% of patients studied so far; is the A3243G mutation of the gene tRNALeu (4,10). In about 10% of the cases, there can be a detection of the variation in RNA T3271C transfer [13]. The percentage of mutated mtDNA in asymptomatic relatives or diabetes, alone, is mild (0-15%) [3]. This defect may be the cause of the deficiency in the activity of the respiratory complexes found in all cases [3]. Recent studies have shown that patients with cardiomyopathy and mitochondrial disease tend to have a more severe clinical course [14]. Holmgren et al. reported different outcomes in patients with MELAS syndrome, with and without cardiomyopathy [14]. Our patient presented growth of the right ventricle without other manifestations. Mortality was 71% in patients with cardiomyopathy and only 26% in those who do not have cardiomyopathy [14]. Parkinsonism as an initial manifestation in MELAS syndrome has seldomly been described [15]. Dystonia, chorea, and myoclonus are more commonly described as initial manifestations. Parkinson`s disease does not present with lactic acidosis as it usually presents in MELAS syndrome [16]. The primary treatment is based on antioxidants, substrates and cofactors of the respiratory chain; however, the results are heterogeneous. Our patient despite treatment with pyridoxine, Coenzyme Q-10, L-arginine had an adverse neurological response with a fatal outcome.
Acknowledgements
The authors want to thanks to Sheyla Rangel Burgos, Dr Rosario Letona.
References
- Scaglia F, Northrop JL (2006) The mitochondrial myopathy encephalopathy, lactic acidosis with stroke-like episodes (MELAS) syndrome: a review of treatment options. CNS Drugs 20: 443-464. [Crossref]
- Marin-Garcia J, Goldenthal MJ (1997) Mitochondrial cardiomyopathy: molecular and biochemical analysis. Pediatr Cardiol 18: 251-260. [Crossref]
- Coelho-Miranda L, Playan A, Artuch R, Vilaseca MA, Colomer J, et al. (2000) Encefalomiopatía mitocondrial, acidosis láctica y accidentes cerebrovasculares (MELAS) en edad pediátrica con la mutación A3243G en el gen del ARNtLeu(UUR) del ADN mitocondrial. Rev Neurol 31: 804-811. [Crossref]
- Goto Y, Nonaka I, Horai S (1990) A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348: 651-653. [Crossref]
- Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP (1984) Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol 16: 481-488. [Crossref]
- Dashe JF, Boyer PJ (1998) Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 39-1998. A 13-year-old girl with a relapsing-remitting neurologic disorder. N Engl J Med 339: 1914. [Crossref]
- Montagna P, Gallassi R, Medori R, Govoni E, Zeviani M, et al. (1988) MELAS syndrome: characteristic migrainous and epileptic features and maternal transmission. Neurology 38: 751-754. [Crossref]
- Ohno K, Isotani E, Hirakawa K (1997) MELAS presenting as migraine complicated by stroke: case report. Neuroradiology 39: 781-784. [Crossref]
- Tarnopolsky M, Stevens L, MacDonald JR, Rodriguez C, Mahoney D, et al. (2003) Diagnostic utility of a modified forearm ischemic exercise test and technical issues relevant to exercise testing. Muscle Nerve 27: 359-366. [Crossref]
- Thomas PK, Griffin JW, Low PA, Poduslo JF (Eds.), Peripheral neuropathy. (3edn), Philadelphia: Saunders; 1984, 1194-218.
- Cano A, Romero AI, Bravo F, Vida JM, Espejo S (2002) Melas syndrome: neuroradiological findings. Radiología 44: 69-74.
- Lau KK, Yang SP, Haddad MN, Butani L, Makker SP (2007) Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes syndrome with hypothyroidism and focal segmental glomerulosclerosis in a paediatric patient. Int Urol Nephrol 39: 941-946. [Crossref]
- Genge A, Massie R, Shefner JM, Dashe JF (2015) Mitochondrial myopathies: Clinical features and diagnosis. UpToDate 2015.
- Holmgren D, Wåhlander H, Eriksson BO, Oldfors A, Holme E, et al. (2003) Cardiomyopathy in children with mitochondrial disease; clinical course and cardiological findings. Eur Heart J 24: 280-288. [Crossref]
- De Coo IF, Renier WO, Ruitenbeek W, Ter Laak HJ, Bakker M, et al. (1999) A 4-base pair deletion in the mitochondrial cytochrome b gene associated with parkinsonism/MELAS overlap syndrome. Ann Neurol 45: 130-133. [Crossref]
- Nakagawa-Hattori Y, Yoshino H, Kondo T, Mizuno Y, Horai S (1992) Is Parkinson's disease a mitochondrial disorder? J Neurol Sci 107: 29-33. [Crossref]