We report a case of paraneoplastic dermatomyositis in a patient with a diagnosis of neuroendocrine tumor of an unknown primary site. A 49-year-old woman, hypertensive, was admitted to our hospital due to a mass detected by abdominal computed tomography, in contact with the liver and gallbladder, having compressive effect on inferior vena cava, and measuring 10.4 x 7.3 x 7.8 cm. Pathological examination showed signet-ring cell neuroendocrine neoplasm with a ki-67 proliferation index of 60%, and microscopic and immunohistochemical findings indicated a metastatic appendiceal goblet cell carcinoid. The association between malignant neoplasms and dermatomyositis has been described in the literature; however, this is the first report of a case of paraneoplastic syndrome associated with a neuroendocrine tumor.
dermatomyositis, paraneoplastic syndrome, neuroendocrine tumor
Paraneoplastic syndromes are a group of rare clinical conditions (1-7.4%) [1], consisting of a set of signs and symptoms that occurs before or simultaneously with solid tumors. As these conditions are involved in tumor progression, they generally improve with cancer treatment. Although the physiopathology of paraneoplastic syndromes has not been fully elucidated, and the conditions have been underdiagnosed, hormonal and humoral factors secreted by neoplastic cells seem to be involved in the pathogenesis of the clinical manifestations [2].
Early recognition of the syndrome is of paramount importance, since it may be the first manifestation of the neoplastic disease. Paraneoplastic syndrome may manifest as signs and symptoms of several chronic inflammatory diseases particularly diffuse connective tissue diseases and dermatopolymyositis (DM). These conditions are commonly present with solid tumors of lung, stomach, prostate, rectum, colon, breast, ovaries and thymus [3].
This report describes the case of paraneoplastic dermatomyositis in a patient with a diagnosis of neuroendocrine tumor of an unknown primary site at the General Hospital of the University of Sao Paulo Medical School, Ribeirão Preto, Brazil.
A 49-year-old woman, with difficult-to-control hypertension, was admitted to the emergency ward presenting abdominal colic pain of severe intensity located in the right hypochondrium. One month later, the abdominal pain had increased, accompanied by chest pain. The initial hypothesis was acute coronary syndrome, which was rejected during hospitalization, and a palpable mass in the right hypochondrium was detected.
After hospital discharge, the patient continued to have severe abdominal pain and progression of tumor growth in right hypochondrium. Total abdominal ultrasound revealed a mass on liver, measuring 7.8 x 7.3 cm. The patient was then referred to the gastric surgery department of this hospital.
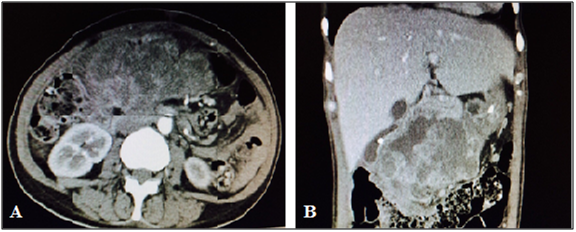
Abdominal computed tomography revealed an irregular mass with heterogeneous enhancement in right hypochondrium, in close contact with the liver and gallbladder, with no cleavage plane with the uncinate process, having a compressive effect on inferior vena cava, measuring 10.4 x 7.3 x 7.8 cm and absence of adenopathy, indicating an expansive formation in the head of pancreas.
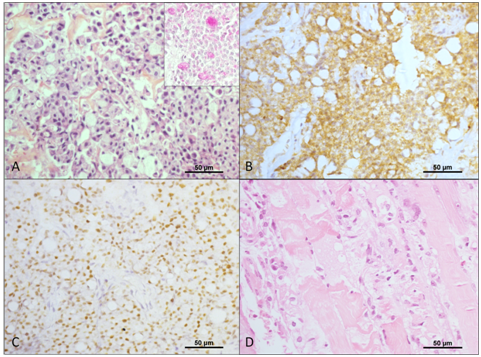
Ultrasound guided biopsy of the lesion yielded signet-ring cell neuroendocrine neoplasm, with a ki-67 proliferation index of 60%, and microscopic and immunohistochemical findings indicated a metastatic appendiceal goblet cell carcinoid.
Upper endoscopy revealed an ulcerative lesion located in the second portion of the duodenum, compatible with tumor invasion by direct extension, which was confirmed by biopsy that showed poorly differentiated small cell malignant neoplasms, with some signet-ring cells.
Other imaging tests were performed (Table 1). Although suggestive of pancreatic neoplasm (and of appendiceal neoplasms by biopsy), the primary site could not be confirmed due to limitations of computed tomography.
Table 1. Imaging tests
|
Description |
Chest computed tomography |
Small right basal pleural reaction, probably benign or as a sequela of the condition. Non-specific pulmonary nodule at left.
|
Abdominal/Pelvic computed tomography |
Extensive, heterogeneous lobulated mass in the head of pancreas/second portion of duodenum, with heterogeneous contrast enhancement, presence of gas and hypodense areas in the interior, measuring 12.6 x 13.3 x 7.3 cm.
|
Upper gastrointestinal endoscopy |
After the passage of endoscope through pylorus, vegetative, friable lesions, with ill-defined margins, areas of ulceration, necrosis, hematin and fibrin were found in the second portion of the duodenum, compatible with duodenal neoplasms.
|
Colonoscopy |
Negative findings on colonoscopy |
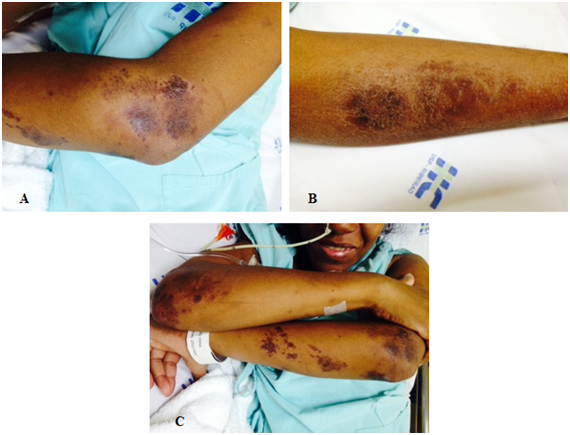
Additionally, the woman presented esophageal dysphagia for solids and liquids, symmetrical proximal muscle weakness, brown-violet lesions on arms and legs, and a high fever (38-39°C) every day. An infectious etiology of fever was ruled out, and a positive response to Naproxen test was indicative of neoplastic fever.
Muscle biopsy of the left biceps showed active inflammatory myopathy, in accordance with laboratory tests (Table 2), hence confirming the diagnosis of paraneoplastic dermatomyositis. The electromyoneurography showed a myopathic pattern compatible with active myositis (rapid repetitive discharges, fibrillation and positive sharp waves in all muscles assessed, with normal motor and sensory conduction).
Table 2. Results of autoantibodies and laboratory tests
|
Results |
Reference values |
Creatine kinase |
4,657 U/L |
24-170 U/L |
Aldolase |
26.0 U/L |
1,0 - 7,6 U/L |
Erythrocyte sedimentation rate |
33.0 mm/h |
< 15 mm/h |
Antinuclear factor |
NR |
NR |
Anti- smooth muscle |
NR |
NR |
Anti-Scl 70 |
16.1 |
< 15.0 |
Anti- SSB |
37.6 |
< 25.0 |
Anti – SSA |
>200 |
< 15.0 |
Anti – Sm |
7.9 |
< 7.0 |
The woman underwent etoposide D1-D3 and cisplatinum (BEP) chemotherapy and the first cycle of immunoglobulin pulse therapy for three days. There was a small improvement of the skin lesions and of muscle weakness. After the second cycle of chemotherapy, the woman experienced febrile neutropenia associated with melena and severe sepsis. After 45 days of hospitalization, she underwent arterial embolization using radio-intervention techniques of tumor cells.
After the third chemotherapy cycle, the woman had significant worsening of status performance, and underwent palliative care. Two months later, she died of severe sepsis secondary to infection of totally implantable venous catheter caused by multi-sensitive S. epidermidis.
Neuroendocrine tumors are a heterogeneous group of tumors with variable biological behavior, histological features and treatment response [4]. Although different histological and anatomical classification systems have been proposed for the neuroendocrine, all of them consider tumor grade (by using the ki-67 proliferation index), and distinction between well-differentiated (indolent tumors) and poorly differentiated (aggressive tumors) tumors [5].
Most tumors are well-differentiated. However, some of them have histological patterns that resemble small-cell poorly differentiated carcinoma, and the diagnosis of neuroendocrine tumors is confirmed by positive immunohistochemical staining for chromogranin and synaptophysin [6].
Despite progresses made in immunohistochemistry for the identification of histological type, etiology and tumor biology, the primary tumor site remains undefined in many cases, and 10-13% of these tumors have signs and symptoms related to the metastatic site [7].
In an epidemiologic study, 35,618 patients with neuroendocrine tumors were identified in the Surveillance, Epidemiology, and End Results in the US population from 1973 to 2004 [8]. The authors reported an increase in the annual incidence from 1.09 per 100,000 in 1973 to 5.25 per 100,000 in 2004. The primary tumor site varies by race, and the lung was the most frequent primary site more among Caucasians, and rectal tumors were more frequent among Asians, Indians and African Americans.
2021 Copyright OAT. All rights reserv
Well-differentiated tumors are classified as having low or intermediate proliferation rate according to the grading system proposed by the World Health Organization for neuroendocrine tumors. The majority of these patients have liver metastases of gastrointestinal origin at diagnosis, some of them neuroendocrine tumors that secrete bioactive substances (p.ex. pheochromocytomas) [9]. The treatment of well-differentiated tumors depends on clinical condition of patients, who can receive either a local therapy (surgical exploration of primary site and/or metastatic lesion) or a systemic therapy (somatostatin analogues or cytotoxic therapy) [9].
High proliferative tumors are a distinct group, with characteristics that are similar to small cell carcinoma of the lung, and mostly treated by platinum containing regimens. A response rate of approximately 66% to cisplatinum and etoposide, that corresponds to the standard therapy [9-11].
In this case reported, the global survival was of eight months, corroborating previous data in the literature reporting a mean global survival of ten months for poorly-differentiated neuroendocrine tumors [8].
When metastases are present at the time when diagnosis is established, the initial evaluation should include chest, abdomen and pelvic imaging tests (computed tomography or magnetic resonance imaging) [12]. Positron emission tomography (PET) is highly sensitive for poorly-differentiated tumors due to their high metabolism rate. On the other hand, octreoscan is relatively insensitive in detecting these tumors and not recommended in this context [12,13]. As there are no significant variations among the therapies based on the primary site of poorly-differentiated metastatic tumor, it is recommended no to postpone the initial treatment prior to detailed investigation
Paraneoplastic syndromes are a group of rare clinical conditions caused by the systemic presence of the tumor. The physiopathology remains unclear, and most cases of paraneoplastic syndromes are underdiagnosed. However, the early recognition of the syndrome is crucial, as it may represent the first tumor-related symptom. In addition, it is worth mentioning that paraneoplastic syndromes manifestations may occur concomitantly or after cancer diagnosis [1].
Inflammatory myopathies are associated with solid neoplasms in a minority of cases. The association between polymyositis and gastric cancer was first described in 1916 [14]. Then a review on 258 cases published in 1976 estimated that the incidence of cancer is seven times higher in patients with dermatomyositis than general population [15]. Other recent population studies have confirmed a higher risk of malignancy among patients with inflammatory myopathies [16-18].
Dermatopolymyositis is a heterogeneous group of multisystem inflammatory myopathies with multiple clinical and laboratory characteristics involving the skin and proximal striated muscle. The prevalence of DM is from 0.5 to 1.0 per 100,000 individuals [19].
Dermatomyositis may be of juvenile or adult type. While juvenile dermatomyositis generally does not manifest as paraneoplastic syndrome, the adult dermatomyositis may be associated with solid neoplasms in 15-20% of cases. For this reason, the screening of patients with clinical characteristics of dermatomyositis for neoplasms is fundamental [19].
The first clinical manifestations of dermatomyositis are symmetrical weakness of proximal muscle, Gottron’s papules, heliotrope rash, photosensitivity, periungueal erythema, and telangiectasias. The diagnosis is confirmed when skin manifestations are associated with the Bohan and Peter criteria that include: (1) symmetrical weakness of proximal muscle; (2) increased skeletal muscle enzyme levels (aldolase, creatine phosphokinase, aspartate transaminase, and alanine transaminase); (3) electromyoneurography compatible with inflammatory myopathy, and (4) active myositis confirmed by muscle biopsy [20].
Patients with DM may have other less frequent symptoms, such as systemic (fever, weight loss, arthralgia), gastrointestinal (malabsorption, intestinal dysmotility), cardiac (atrial and ventricular tachyarrhythmia, dilated cardiomyopathy), pulmonary (pulmonary interstitial disease, dyspnea) and vascular (Raynaud's phenomenon, vasculitis) symptoms [19]. The presence of pulmonary interstitial disease decreases the risk of neoplasms, for yet unknown reasons [21].
The patient of this case report presented signs and symptoms characteristic of DM, such as hyperchromic skin lesions, symmetrical weakness of proximal muscle, and biochemical changes, including increased muscle enzymes (creatine phosphokinase: 4,657 U/L and aldolase: 26.0 U/L), electromyography compatible with inflammatory myositis, and active inflammatory myopathy, evidenced by muscle biopsy. Patient’s advanced age (greater than 40 years), increased creatine phosphokinase levels and absence of pulmonary interstitial disease strengthen the association between DM and cancer.
Neoplasms relative risk is around 26 per 1,000,000. It increases in the first five years following the diagnosis of DM, and the incidence peak occurs within two years of diagnosis [22]. Requena et al. demonstrated that the time lapse from DM diagnosis to cancer (or recurrence) varied by 20 months (mean of 7 months) [22]. Buchbinder et al. reported a frequency of 36% of simultaneous diagnosis of neoplasms (within seven days before and after the inflammatory myopathy diagnosis). In this case report, DM symptoms were experienced concomitantly the diagnosis of neuroendocrine tumor, and approximately two months following the detection of abdominal mass.
Although some DM cases may be diagnosed at tumor recurrence, other previously diagnosed cases may present recurrence of myopathy with solid neoplasms. Neoplasms more frequently related with DM are breast cancer and gynecological tumor in women, lung cancer in men and gastrointestinal cancer in both sexes. The risk is substantially greater in the elderly than in male patients [24,25]. There is no data in the literature on neoplastic DM associated with neuroendocrine tumors, or with hidden primary tumors of other histologies.
Clinical manifestations of paraneoplastic syndromes are generally controlled with the therapy implemented, with complete remission in cases of objective tumoral response. Recurrence of inflammatory myopathy symptoms have been reported [26], and the presence of neoplasms seem to not influence the severity of DM clinical manifestations.
In cases when oncologic therapy is not possible, when the complete response is not achieved, or when the onset of anti-neoplastic therapy is delayed for some reason, administration of oral corticosteroid is indicated [23].
In this case reported, due to the severity of symptoms, the cytotoxic therapy combined with pulse therapy was the treatment of choice, leading to a partial improvement of symptoms right after the first therapy cycle.
Despite progresses made in oncology, prognosis of neuroendocrine tumors with high proliferation rate remains guarded. Paraneoplastic syndromes are still underdiagnosed, as they precede clinical manifestations of neoplasms.
This case report described a favorable clinical course, and death of the patient within months of the diagnosis, indicating the aggressive course of poorly-differentiated metastatic neuroendocrine carcinomas. The associated inflammatory myopathy made this patient more susceptible to infections, with systemic repercussions and unfavorable evolution.
- Chapireau D, Adlam D, Cameron M, Thompson M (2010) Paraneoplastic syndromes in patients with primary oral cancers: a systematic review. Br J Oral Maxillofac Surg 48: 338-344. [Crossref]
- Tarantino AB, Silveira IC, Blundi E (1980) Síndromes Paraneoplásicas. JBM 38: 43-54.
- Marques Neto JF (1992) Sídnromes paraneoplásicas: revisão das manifestações osteomioarticulares. Rev Cienc Med, Campinas 1: 49-53.
- Wang SC, Parekh JR, Zuraek MB, Venook AP, Bergsland EK, et al. (2010) Identification of unknown primary tumors in patients with neuroendocrine liver metastases. Arch Surg 145: 276-280. [Crossref]
- Klimstra DS, Modlin IR, Coppola D, Lloyd RV, Suster S (2010) The pathologic classification of neuroendocrine tumors: a review of nomenclature, grading, and staging systems. Pancreas 39: 707-712. [Crossref]
- Catena L, Bichisao E, Milione M, Valente M, Platania M, et al. (2011) Neuroendocrine tumors of unknown primary site: gold dust or misdiagnosed neoplasms? Tumori 97: 564-567. [Crossref]
- Spigel DR, Hainsworth JD, Greco FA (2009) Neuroendocrine carcinoma of unknown primary site. Semin Oncol 36: 52-59. [Crossref]
- Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, et al. (2008) One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 26: 3063-3072. [Crossref]
- Pavel M, Baudin E, Couvelard A, et al. (2012) ENETS Consensus Guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology 2012; 95: 157. [Crossref]
- Moertel CG, Kvols LK, O'Connell MJ, Rubin J (1991) Treatment of neuroendocrine carcinomas with combined etoposide and cisplatin. Evidence of major therapeutic activity in the anaplastic variants of these neoplasms. Cancer 68: 227-32. [Crossref]
- Hainsworth JD, Spigel DR, Litchy S, Greco FA (2006) Phase II trial of Paclitaxel, carboplatin and etoposide in advanced poorly differentiated neuroendocrine carcinoma: a Minnie Pearl Cancer Research Network Study. J Clin Oncol 24: 3548. [Crossref]
- Hicks RJ (2010) Use of molecular targeted agents for the diagnosis, staging and therapy of neuroendocrine malignancy. Cancer Imaging 10 Spec no A: S83-91. [Crossref]
- Strosberg JR, Coppola D, Klimstra DS, Phan AT, Kulke MH, et al. (2010) The NANETS consensus guidelines for the diagnosis and management of poorly differentiated (high-grade) extrapulmonary neuroendocrine carcinomas. Pancreas 2010; 39: 799. [Crossref]
- Stertz O (1916) Polymyositis. Berl Klin Wochenschr 53: 489.
- Barnes BE, Mawr B (1976) Dermatomyositis and malignancy. A review of the literature. Ann Intern Med 84: 68-76. [Crossref]
- Sigurgeirsson B, Lindelöf B, Edhag O, Allander E (1992) Risk of cancer in patients with dermatomyositis or polymyositis. A population-based study. N Engl J Med 326: 363-367. [Crossref]
- Buchbinder R, Forbes A, Hall S, Dennett X, Giles G (2001) Incidence of malignant disease in biopsy-proven inflammatory myopathy. A population-based cohort study. Ann Intern Med 134: 1087-1095. [Crossref]
- Stockton D, Doherty VR, Brewster DH (2001) Risk of cancer in patients with dermatomyositis or polymyositis, and follow-up implications: a Scottish population-based cohort study. Br J Cancer 85: 41-45. [Crossref]
- Dalakas MC, Hohlfeld R (2003) Polymyositis and dermatomyositis. Lancet 362: 971-982. [Crossref]
- Bohan A, Peter JB (1975) Polymyositis and dermatomyositis (first of two parts). N Engl J Med 292: 344-347. [Crossref]
- Norrenberg S, Gangji V, Del Marmol V, Soyfoo MS (2012) Diffuse muscular pain, Skin tightening and nodular regenerative hyperplasia revealing paraneoplastic amyopathic Dermatomyositis due to testicular câncer. Case reports in Reumatology. [Crossref]
- Requena C, et al. (2014) Dermatomiositis paraneoplasica: studio de 12 casos. Actas Dermosifiliogra.
- Landriscina M, Gerardi AM, Fersini A, Modoni S, Stoppino LP, et al. (2013) Multiple skeletal muscle metastases from colon carcinoma preceded by paraneoplastic dermatomyositis. Case Rep Med 2013: 392609. [Crossref]
- Wada C, Hua CN, Carney ME (2014) Paraneoplastic syndrome in Hawai’i: a case of Dermatomyositis associated with endometrial cancer. Hawai J Med Public Health 73: 112-4. [Crossref]
- Kaltsas GA, Besser GM, Grossman AB (2004) The diagnosis and medical management of advanced neuroendocrine tumors. Endocr Rev 25: 458-511. [Crossref]
- Manchul LA, Jin A, Pritchard KI, Tenenbaum J, Boyd NF, et al. (1985) The frequency of malignant neoplasms in patients with polymyositis-dermatomyositis. A controlled study. Arch Intern Med 145: 1835-1839. [Crossref]