Abstract
Nonalcoholic fatty pancreas disease (NAFPD) is closely linked to nonalcoholic steatohepatitis (NASH), suggesting that the two conditions share a common etiopathogenetic background. In Addition, growing evidence indicates that endogenous ethanol (EE) plays a fundamental role in NASH pathogenesis. Accordingly, it is intuitively appealing to assume that EE plays also a causative role in NAFPD development. This connection is further supported by the finding that NAFPD shares with alcoholic fatty pancreas disease (AFPD) similar metabolic signaling pathways and histopathological features. However, low blood alcohol concentrations (BAC) along with the alleged inability of gut microbiota to produce toxic amounts of ethanol are the main obstacles to validate the idea of an endogenous alcoholic fatty pancreas disease (EAFPD). Here, we provide a mechanistic explanation reconciling the EAFPD hypothesis with these apparently conflicting observations. The key conclusions of our investigation are as follows. First, ethanol is a prodrug, implicating that under extensive presystemic metabolism BACs can be low and/or absent. Second, oro-gastrointestinal microbiota may produce higher amounts of ethanol than those required to cause AFPD. Last, livers of NASH/NAFPD individuals overexpress all genes encoding alcohol-metabolizing enzymes identically to livers of patients with alcoholic hepatitis. Even more importantly, the up-regulation of these genes is higher in the early steatotic stage of nonalcoholic fatty liver disease than in alcoholic hepatitis. This suggests a greater exposure of the liver, and by extension, of the pancreas, to ethanol in the former than in the latter condition. In summary, this paper provides a mechanistic framework for how NAFPD indeed may be an EAFPD.
Key words
acetaldehyde, alcoholic fatty pancreas disease, endogenous alcoholic fatty pancreas disease, endogenous ethanol, fatty acid ethyl esters, nonalcoholic fatty pancreas disease
Introduction
Nonalcoholic fatty pancreas disease (NAFPD) is a broad-spectrum disorder ranging from simple pancreatic steatosis to acute and chronic fibrosing nonalcoholic steatopancreatitis (NASP) and pancreatic cancer [1–4]. There are two distinct forms of this disease: primary or obesity-associated NAFPD, and secondary forms, which are linked to congenital syndromes, toxic agents, medications, viruses, and severe malnutrition [5]. Its risk factors include insulin resistance, prediabetes, diabetes, hyperferritinemia, central obesity, and hypertriglyceridemia [6,7]. Additionally, body mass index, HbA1c, total cholesterol, LDL-cholesterol, platelet count and systolic blood pressure are also a putative risk for NAFPD [8]. Based on these findings, it is clear that NAFPD and NAFLD share very similar environmental-related risk factors, suggesting a common etiological background between them. Agreeing with this observation, hepatic steatosis increases by nearly 14-fold the odds of having NAFPD [9], whereas ~70% of the individuals with NAFLD concurrently had NAFPD [10]. Moreover, interlobular and total pancreatic fat are positively correlated with NAFLD activity score [11], whereas pancreatic intralobular fat and impaired pancreatic β-cell function are heralds of NASH [11,12].
It is known that NAFPD shares with alcoholic fatty pancreas disease (AFPD) similar metabolic pathways [3,13–16], as well as histopathological features [1,3,4,17–25]. In addition, NAFLD patients produce significantly more endogenous ethanol (EE) than controls, suggesting that it plays an important role in its pathogenesis [26–32]. Thus, as NAFPD is closely linked to NAFLD and NAFLD-related conditions [1,5,10,33–35], it follows that EE can also exert a relevant role in the pancreatic injury. Taken together, these data allow us to hypothesize that NAFPD and AFPD are the same disease with different clinical presentations.
Currently, there is no study investigating a causal relationship between EE and NAFPD. Hence, the main focus this article is to provide a mechanistic explanation of how NAFPD may indeed be an endogenous alcoholic fatty pancreas disease (EAFPD). In this context, we will demonstrate how subjects with NAFLD/NAFPD can produce higher amounts of ethanol than those required for causing AFPD [36] without inducing significant blood alcohol concentration (BAC).
The hypothesis
Ethanol is a prodrug
Similarly to other prodrugs, ethanol must be metabolized to acetaldehyde (ACD) and/or fatty acid ethyl esters (FAEEs) for exerting the most of its injurious effects. Compelling evidence comes from the finding that inhibitors of oxidative and nonoxidative metabolism of ethanol abrogate its cytotoxic effects [37–43]. The obvious implication of this is that an endogenous alcoholic disease may develop concomitantly with low/negligible BACs.
Oro-gastrointestinal production of ethanol and acetaldehyde
Gut microbiota of healthy individuals produces trace quantities of ethanol from unabsorbed dietary carbohydrates [44–47]. Next, it is converted in the liver to acetaldehyde, which in turn is oxidized to non-toxic concentrations of acetate [47]. The demonstration that the baker’s yeast elicits a 4-fold increase in gastric EE concentration supports this view. Further, pharmacological blockade of alcohol dehydrogenase (ADH) elicits a 130-fold rise in EE content of hepatic venous blood in rats [47].
On the other hand, in patients with NAFLD/NAFPD the emergence of small intestinal bacterial overgrowth (SIBO) [48–53] is associated with significantly increased body concentrations of EE compared to controls [27,32,54,55]. Further, concurrent obesity-related hypochlorhydric/achlorhydric conditions may also contribute to EE production [56–61]. Thereby, in dysbiotic disorders gut concentrations of EE may be even higher than those found after moderate drinking [62–64]. In line with this, genomic studies suggest that alcohol-metabolizing enzymes are differentially expressed in livers of NAFLD/NAFPD patients [28,30,65–68]. These up-regulated enzymes can work at 500 to 3000 mM (~23 to 138 g) ethanol concentrations [69].
Of note, gut microbial makeup of these dysbiotic conditions includes both alcohol-producing [26,51,62,64,70–79] and alcohol-degrading microorganisms [62,80–85]. In this setting, both host and microbial enzymes of the oro-gastrointestinal tract produce ethanol and oxidize it into acetaldehyde in a dose-dependent manner [70,83,86–91]. Interestingly, on the one hand, exposure to ethanol stimulates alcohol-degrading enzymes [67,92], and on the other, it inhibits the catalytic action of aldehyde dehydrogenase (ALDH), thereby contributing to acetaldehyde accumulation [93–98]. This, coupled with hepatic first-pass metabolism (FPM) of ethanol may reduce substantially BACs. A study has clearly demonstrated this extensive metabolism of ethanol in chronic alcohol abusers. According to it, to maintain a steady-state BAC between 1.5 to 4.5 mg/dL, it was necessary performing a constant intravenous infusion of ethanol at a rate of 57.6 to 115.2 g/d [99]. In this setting, blood ethanol diffuses rapidly into liver and gastrointestinal tract, where it undergoes extensive first-pass metabolism by both host and microbial enzymes. For comparison purposes, mean BAC was 7.1 mg/dL 12-h after an overnight fast in obese patients with NAFLD [32]. For maintaining this BAC level, each patient would have to produce 114 g of EE or receiving continuous intravenous ethanol infusion at a rate of ~9.5 g/h.
A remarkable exception to this metabolic pattern is frequently observed in the auto-brewery syndrome. This disorder, in addition to SIBO, is accompanied by fungal overgrowth of alcohol-producing organisms such as Candida sp and/or Saccharomyces cerevisiae [62,71,72,76,100–102]. Characteristically, auto-brewery syndrome patients produce massive quantities of EE from dietary carbohydrates. In this scenario, the first-pass metabolism of ethanol is insufficient to avoid drunkenness and BACs can reach 250-350 mg/dL [76].
Endogenous production of fatty acid ethyl esters (FAEE)
Presently, there is no study investigating the role of FAEEs in NASH/NAFPD pathogenesis. Notwithstanding, it is known that healthy subjects not exposed to exogenous ethanol have endogenous trace of FAEEs in body fluids [103,104]. As the two substrates required for FAEEs biosynthesis, ethanol [68][77] [105] and free fatty acids [13,106–109], are significantly elevated in NASH/NAFPD body fluids, it is quite possible they are formed in toxic amounts in the pancreas and oro-gastrointestinal tract.
Harmful effects of acetaldehyde and fatty acid ethyl esters on the pancreas
The ALDH activity in the pancreas is nearly fivefold lower than that of the liver, contributing in this way to acetaldehyde glandular accumulation [110]. Despite this fact, data from animal studies have shown that acetaldehyde by itself does not elicit directly acute pancreatitis [111,112]. Conversely, some studies support the involvement of acetaldehyde in both pancreatic fibrosis and carcinogenesis [16,113–117]. Dysbiotic gut microbiota is able to convert both exogenous and endogenous ethanol to acetaldehyde in a dose-dependent manner [70]. This extrahepatic acetaldehyde (generated outside the liver) is about 30 to 330-fold more hepatotoxic than that formed in the liver [118,119]. Accordingly, it is reasonable to assume that extrahepatic acetaldehyde may play a protagonist role in more advanced stages of NAFPD pathogenesis.
On the other hand, the critical role of FAEEs in acute pancreatic injury is already well-established [43,120–122]. In keeping with these data, the pharmacological blockage of FAEEs biosynthesis prevents [43,120], whereas the stimulation of its production promotes acute pancreatic damage [43,120,121]. By contrast, the role of FAEEs in pancreatic fibrosis is still far from being fully settled [123].
Estimating daily endogenous ethanol production and its first-pass metabolism
For the calculations, we use a validated physiologically-based pharmacokinetic model of body alcohol metabolism [124] and demographic data of patients taken from a published study [32]. According to this study, NAFLD patients had an average BAC of 7.14 mg/dL 12-h after an overnight fast [32]. We assume that the mean patient height was 1.74 m (68.8976 inches), and Watson’s equation was used for total-body water calculation (TBW) [124].
Equation (1): calculating TBW
ΣVd = 2.44 – (0.09516 x age) + [0.1074 x (height in inches) x 2.54)] + [0.3362 x weight in pounds / 2.2045)]
ΣVd is TBW or volume of distribution in which alcohol will be dispersed according to the age, weight, height, and patient gender [124]. Inserting the figures from Menezes et al’s study [32] (age, 48 years, BMI, 35 kg/m², and body weight, 107.20 Kg [236.3355 lb]), we obtain:
ΣVd = [2.44 – (0.09516 x 48)] + [0.1074 x (68.8976 x 2.54)] + [(0.3362 x 236.3355) /2.2045] = 52.70 liters. Then, we use the modified Widmark’s equation.
Equation (2): calculating total alcohol consumed/produced (TAC)
TAC = ΣVd x (BACobj + β1n x t) / BℓH2O
TAC means total alcohol endogenously produced; BACobj is the objective blood-alcohol concentration result; β1n is the range of the ethanol elimination rate (10 to 20 mg/dL/h for healthy individuals and 20 to 30 mg/dL/h for heavy drinkers); t is the time from the start of drinking (here meaning the start of carbohydrate intake) until the time of the BAC test, and BℓH2O is the constant (80.65) approximate percentage of water in blood. It follows that TAC = 52.70 x (7.14 + 20 x 12)/80.65 = 161.49 g of ethanol [124].
This means each patient produces 161.49 g of ethanol after a12-h overnight fast (Figure 1). Hence, by extrapolation, the daily production of ethanol should reach 484 g after eating three equicaloric meals (3 x 161g = 484g). Once the patients’ BAC is consistently low, one concludes that ethanol has undergone extensive conversion to acetaldehyde in the gut-liver axis. The fist-pass metabolism (FPM) of ethanol in the gut-liver axis can also be accurately calculated. For this, we need initially to calculate the alcohol burden in the circulation utilizing some data we already described.
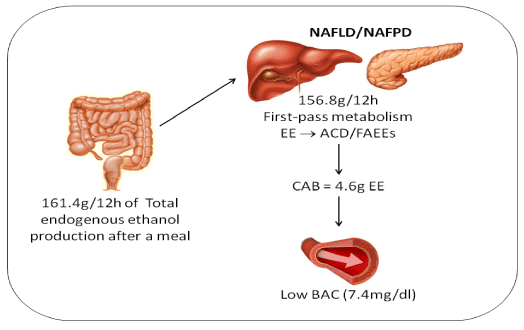
Figure 1. Estimate of the production, metabolism, and circulation of endogenous ethanol according to a physiologically-based pharmacokinetic model of body alcohol metabolism. The calculations are based in 20 NAFLD patients with the following average values: age: 48 years, BMI: 35 kg/m², height: 1.74m, weight: 107.2 Kg, and BAC: 7.1 mg/dL 12-h after an overnight fast [32]. The activity of aldehyde dehydrogenase (ALDH) in the pancreas is nearly fivefold lower than that in the liver, whereas non-oxidative metabolism of ethanol is 10 times higher in the pancreas than in the liver, contributing to accumulation of toxic acetaldehyde and fatty acid ethyl esters in the gland. EE = Endogenous Ethanol; ACD = Acetaldehyde; FAEEs = Fatty Acid Ethyl Esters; CAB = Circulating Alcohol Burden; BAC = Blood Alcohol Concentration.
Equation (3): calculating circulation alcohol burden (CAB).
CAB = (BACobj x ΣVd) / 80.65. Inserting the data into the equation, one obtains CAB = (7.14 x 52.70) / 80.65 = 4.60 g of ethanol.
FPM of ethanol can be estimated subtracting circulating alcohol burden (4.60 g) from total alcohol produced (161.49 g). We obtain the amount of ethanol metabolized (161.49-4.60 = 156.80 g) (Figure 1) [124]. This is consistent with the finding that blind-loop contents of a 0.3 kg rat oxidize ethanol at a rate of 123 mg/h [70]. If these data could be extrapolated to a SIBO patient weighing 107.20 kg, FPM of EE should reach about 43.90 g/h.
Supporting these observations, gene of all alcohol-metabolizing enzyme are differentially expressed in livers of NASH patients [30]. Also, intragastric administration of ethanol (1 g/kg) to rats with SIBO evoked a 10-fold increase in the portal concentration of acetaldehyde, whereas portal and systemic BACs reached negligible values [70]. Another study showed similar portal venous concentrations of ethanol in obese and lean mice, suggesting intraluminal FPM of ethanol in the obese group [125]. In addition, host ADH3 present in the stomach, small bowel, and liver may play a significant role in FPM of ethanol. It works at ethanol concentrations of 0.5 to 3 M (~23-138 g) [126]. Ultimately, adult human liver can metabolize ~240 g of ethanol per day [127,128].
Hepatic overexpression of genes encoding alcohol-metabolizing enzymes
Genomic studies demonstrated that all genes involved in oxidative metabolism of ethanol and acetaldehyde are up-regulated in the livers of NAFLD/NASH patients [28,30,65,66,68]. This suggests that the liver and, probably the pancreas, of these patients consistently metabolize high amounts of EE.
Discussion
Since ethanol is a prodrug, it becomes clear that an endogenous alcoholic disease may develop with low or even undetectable BACs. Yet, although NAFPD has the same histopathological features and metabolic pathways of AFPD, it is not recognized as an EAFPD. The underlying rationale for this is the purported inability of gut microbiota to produce toxic amounts of ethanol and the low BACs found in NAFLD/NAFPD [70,129].
As opposed to this, our calculations showed that an obese patient with NAFLD may produce larger amounts of ethanol than those required for causing AFPD [36] in the presence of low BAC. The finding that gene expression of alcohol-metabolizing enzymes in NAFLD livers is indistinguishable from that of alcoholic hepatitis supports this view [68]. Notably, hepatic ADH4 expression was shown to be ~40-fold higher in NASH livers than in controls [28]. This means that hepatic concentrations of EE can reach up to 1.5 M (6.9 g/dL) without enzyme saturation [69]. Even more surprisingly was the finding that the expression of alcohol-metabolizing genes was higher in livers with mild NAFLD than in alcoholic hepatitis [68].
It is known that as soon as EE is formed, it is dose-dependently converted to acetaldehyde and/or FAEEs. To illustrate, gut contents of rats with SIBO oxidizes ethanol at a rate of 0.99 to 1.99 µM/min/ml, respectively, in anaero- and aerobiosis [70]. Thus, a NAFPD patient with a dysbiotic small bowel contents around 2000 ml could metabolize ethanol at a rate of 3980 µM/min. This amounts to an ethanol elimination rate of 528 g per day, without including gastric, colonic, and hepatic FPM. This key role of gut microbiota in FPM of ethanol is supported by studies showing that oral antibiotics protect against both NAFLD/NAFPD [130–134] and alcoholic liver disease [135]. Furthermore, germ-free animals are also protected against diet-induced obesity-related disorders [136–138] as well as ethanol-induced liver injury [139].
Besides, individuals chronically exposed to ethanol convert it to its metabolites at a rate 3 to 4 fold higher than that of healthy controls [124,140]. A plausible explanation is that chronic exposure to ethanol leads to hyperinsulinemia [125], which shifts energy supply from glucose to ketone bodies [65]. In turn, high ketone bodies alongside oxidized carbohydrates and iron overload shift ethanol metabolism from low Km ADH1 (that works in the millimolar range) to high Km alcohol-metabolizing enzymes (that work in the molar range) [65,141] such as cytochrome P450 2E1 (CYP2E1), ADH4, and ADH3 [28,30,66,69,126,142,143]. The net result is that large amounts of ethanol may cause organ damage concurrently with negligible BACs (Figure 2) [28,30,65–68].
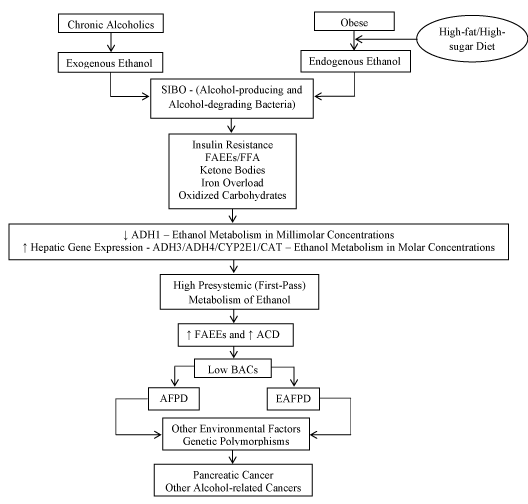
Figure 2. Possible common pathways in the pathophysiology of exogenous and endogenous Alcoholic Fatty Pancreas Disease. Chronic alcoholics and obese individuals are respectively exposed to high concentrations of exogenous and endogenous ethanol formed from dietary carbohydrates. This leads to SIBO rich in both alcohol-producing and alcohol–degrading microorganisms. Then, IR and hyperinsulinemia shift energy supply from glucose to ketone bodies. Ketones along FFA, FAEEs, Iron Overload, and Oxidized Carbohydrates inhibit low Km ADH1 activity and induce gene expression of high Km ADH3, ADH4, CYP2E1, and Catalase activities. As a result, the ability to metabolizing ethanol is shifted from millimolar to molar range. The extensive presystemic (first-pass) metabolism of molar quantities of ethanol generates ACD/FAEEs, leading to AFPD/EAFPD and to the consequent insignificant BACs. Other environmental factors such as obstructive sleep apnea, LPS, glyceraldehyde, methylglyoxal, AGEs, RAGEs, nitric oxide/peroxynitrite, and ALEs may also contribute to both AFPD and EAFPD development. Importantly, genetic polymorphisms of alcohol-metabolizing enzymes as well as genetic mutations specifically linked to chronic pancreatitis such as CFTR, SPINK1, PRSS1, (CTR)C, and (CLDN)2 confer increased risk of developing disease progression and pancreatic cancer. Of note, obese individuals with NAFPD share with alcoholics the same increased risk of alcohol-related cancers, reinforcing the hypothesis that they are also exposed to abnormally high concentrations of endogenous ethanol. SIBO = Small Intestinal Bacterial Overgrowth; IR = Insulin Resistance; FAEE = Fatty Acid Ethyl Ester; FFA = Free Fatty Acid; ADH = Alcohol Dehydrogenase; CYP2E1 = Cytochrome P-450 2E1; CAT = Catalase; ACD = Acetaldehyde; BAC = Blood Alcohol Concentration; AFPD = Alcoholic Fatty Pancreas Disease; EAFPD = Endogenous Alcoholic Fatty Pancreas Disease; Km = Michaelis Constant; LPS = Lipopolysaccharide (Endotoxin); AGE = Advanced Glycation End Products; RAGE = Receptor for Advanced Glycation End Products; ALE = Advanced Lipid Peroxidation End Products; CFTR = Cystic Fibrosis Transmembrane Conductance Regulator; SPINK1 = Kazal-type Serine Protease Inhibitor-1; PRSS = Protease Serine 1; (CTR)C = Chymotrypsin C; (CLDN)2 = Claudin 2;
Big differences between BACs and orally administrated ethanol in healthy individuals are found only when it is ingested with a large meal [144]. It may delay for several hours gastric delivery of ethanol to small bowel absorption [145,146]. Seen from this perspective, the pharmacokinetics of EE in patients with NAFPD is likely even more erratic than the above-mentioned.
Unlike chronic alcoholics, NAFPD patients endogenously and gradually produce ethanol from dietary carbohydrates over meals instead of ingesting it. Moreover, both EE production and metabolization are dose-dependent phenomena which happen simultaneously. Lastly, delayed gastric emptying [147] coupled with SIBO-induced prolonged small bowel transit time [148] allow efficient intraluminal production and metabolization of molar amounts of ethanol. In this way, intraluminal ethanol cannot reach saturating concentrations as well as arrive at the liver in saturating quantities [144]. This process is made easier by the fact that increased concentrations of ACD reduce portal blood flow, causing a decrease in ethanol absorption and, consequently lower portal and systemic BACs [149,150]. This is consistent with a study in chronic alcoholics in which to maintain a steady-state BAC between 1.5 to 4.5 mg/dL, it was necessary performing a constant intravenous infusion of ethanol at a rate of 57.6 to 115.2 g/d [99]. Briefly, our analysis suggests that the ability of obese patients with NAFLD/NAFPD and SIBO to produce and metabolize ethanol is greater than that of chronic alcoholics.
The non-oxidative metabolism of ethanol is about 10 times higher in the pancreas than in the liver [151], whereas pancreatic ability for oxidizing ethanol is relatively low [16], favoring net build-up of FAEEs within the gland. In addition, chronic alcohol exposure induces overexpression of FAEE synthase-related genes in the pancreas and liver [152]. Thus, given the evidence that FAEEs are synthesized by individuals not exposed to exogenous ethanol [103,104] and that NAFLD/NAFPD subjects are exposed to high concentrations of EE [26–32,65–68], it is unsurprising that FAEEs play a causative role in NAFPD pathogenesis. It is worth mentioning that the pharmacologic blockage of the hydrolysis of FAEEs to free fatty acids prevents pancreatic necrosis [153]. So, FAEEs are sufficient but not necessary for pancreatic injury. Given that free fatty acids are significantly increased in NAFPD/NASH, it is plausible that they may also mediate the pancreatotoxic effects of EE [13,106–109].
Several genetic polymorphisms, including those of alcohol-metabolizing enzymes, are positively associated with an increased occurrence of chronic alcoholic pancreatitis [154–158]. Based on this and in the chronic long-term exposure to EE, we postulate that NAFPD patients carrying these polymorphisms are at increased risk of developing disease progression [159,160] and alcohol-related cancers [61,160–164].
Our analysis indicates that together, endogenous acetaldehyde and FAEEs/free fatty acids may recapitulate the full clinical-pathologic spectrum of NAFPD. Yet, we cannot rule out the involvement of others toxic compounds, particularly during NAFPD progression. Indeed, some studies suggest a possible causal relationship between NAFLD/NAFPD-related disorders with chronic intermittent hypoxia elicited by obstructive sleep apnea [165,166], bacterial endotoxin [167–169], glyceraldehyde [170], methylglyoxal [171–173], advanced glycation end products (AGE)s [174], receptor for advanced glycation end products (RAGE) [175], nitric oxide/peroxynitrite [176], and lipid peroxidation end products [177,178].
Conclusion
In conclusion, seen from this perspective becomes easier to understand how huge amounts of ethanol (ingested or endogenously produced) may elicit organ injury with insignificant BACs. The EAFPD hypothesis reconciles the apparently contradictory development of an endogenous alcoholic disease concomitantly with low BACs. It can be easily tested in both humans and animal models by conventional laboratorial and histopathological technics. Its main limitation is that much of the supporting evidence comes from in vitro, observational, and preclinical studies.
Ultimately, NAFPD is a worldwide public health problem whose causative agents remain to be identified. Therefore, it is expected that if the EAFPD hypothesis is validated by further studies, it might help to implement effective preventive and therapeutic approaches.
References
- Smits MM, van Geenen EJ (2011) The clinical significance of pancreatic steatosis. Nat Rev Gastroenterol Hepatol 8: 169-177. [Crossref]
- Rajesh G, Kumar H, Menon S, Balakrishnan V (2012) Pancreatitis in the setting of the metabolic syndrome. Indian J Gastroenterol 31: 79-82. [Crossref]
- Yan MX, Ren HB, Kou Y, Meng M, Li YQ (2012) Involvement of nuclear factor kappa B in high-fat diet-related pancreatic fibrosis in rats. Gut Liver 6: 381-387. [Crossref]
- Tomita Y, Azuma K, Nonaka Y, Kamada Y, Tomoeda M, Kishida M, et al. (2014) Pancreatic Fatty degeneration and fibrosis as predisposing factors for the development of pancreatic ductal adenocarcinoma. Pancreas 43: 1032–1041. [Crossref]
- Prachayakul V, Aswakul P (2015) Pancreatic Steatosis: What Should Gastroenterologists Know? JOP J Pancreas 16: 227–231.
- Ou HY, Wang CY, Yang YC, Chen MF, Chang CJ (2013) The association between nonalcoholic fatty pancreas disease and diabetes. PLoS One 8: e62561. [Crossref]
- Wong VW, Wong GL, Yeung DK, Abrigo JM, Kong AP, Chan RS, et al. (2014) Fatty Pancreas, Insulin Resistance, and ß-Cell Function: A Population Study Using Fat-Water Magnetic Resonance Imaging. Am J Gastroenterol 109: 589-597. [Crossref]
- Wu WC, Wang CY (2013) Association between non-alcoholic fatty pancreatic disease (NAFPD) and the metabolic syndrome: case-control retrospective study. Cardiovasc Diabetol 12: 77.
- Al-Haddad M, Khashab M, Zyromski N, Pungpapong S, Wallace MB, et al. (2009) Risk factors for hyperechogenic pancreas on endoscopic ultrasound: a case-control study. Pancreas 38: 672-675. [Crossref]
- Lee JS, Kim SH, Jun DW, Han JH, Jang EC, et al. (2009) Clinical implications of fatty pancreas: correlations between fatty pancreas and metabolic syndrome. J Clin Biochem Nutr 46: 1869–1875.
- van Geenen EJ, Smits MM, Schreuder TC, van der Peet DL, Bloemena E, et al. (2010) Nonalcoholic fatty liver disease is related to nonalcoholic fatty pancreas disease. Pancreas 39: 1185-1190. [Crossref]
- Siddiqui MS, Cheang KL, Luketic VA, Boyett S, Idowu MO, et al. (2015) Nonalcoholic Steatohepatitis (NASH) Is Associated with a Decline in Pancreatic Beta Cell (β-Cell) Function. Dig Dis Sci 60: 2529-2537. [Crossref]
- Zyromski NJ, Mathur A, Gowda GAN, Murphy C, Swartz-Basile DA,Wade TE, et al. (2009) Nuclear magnetic resonance spectroscopy-based metabolomics of the fatty pancreas: implicating fat in pancreatic pathology. Pancreatology 9: 410–419.
- Zhao ZZ, Xin LL, Xia JH, Yang SL, Chen YX, Li K (2015) Long-term High-fat High-sucrose Diet Promotes Enlarged Islets and ß-Cell Damage by Oxidative Stress in Bama Minipigs. Pancreas 44: 688–695.
- Zhou X, Han D, Xu R, Li S, Wu H, Qu C, et al. (2014) A Model of Metabolic Syndrome and Related Diseases with Intestinal Endotoxemia in Rats Fed a High Fat and High Sucrose Diet. PloS One 9: e115148. [Crossref]
- Clemens DL, Wells MA, Schneider KJ, Singh S (2014) Molecular mechanisms of alcohol associated pancreatitis. World J Gastrointest Pathophysiol 5: 147-157. [Crossref]
- Mathur A, Marine M, Lu D, Swartz-Basile DA, Saxena R, et al. (2007) Nonalcoholic fatty pancreas disease. HPB (Oxford) 9: 312-318. [Crossref]
- Fullenkamp AM, Bell LN, Robbins RD, Lee L, Saxena R, et al. (2011) Effect of different obesogenic diets on pancreatic histology in Ossabaw miniature swine. Pancreas 40: 438-443. [Crossref]
- Matsuda A, Makino N, Tozawa T, Shirahata N, Honda T, et al. (2014) Pancreatic fat accumulation, fibrosis, and acinar cell injury in the Zucker diabetic fatty rat fed a chronic high-fat diet. Pancreas 43: 735-743. [Crossref]
- Yan M-X, Li Y-Q, Meng M, Ren H-B, Kou Y (2006) Long-term high-fat diet induces pancreatic injuries via pancreatic microcirculatory disturbances and oxidative stress in rats with hyperlipidemia. Biochem Biophys Res Commun 347: 192–199. [Crossref]
- Ida S, Ohmuraya M, Hirota M, Ozaki N, Hiramatsu S, et al. (2010) Chronic pancreatitis in mice by treatment with choline-deficient ethionine-supplemented diet. Exp Anim 59: 421-429. [Crossref]
- De Angelis C, Valente G, Spaccapietra M, Angonese C, Del Favero G, et al. (1992) Histological study of alcoholic, nonalcoholic, and obstructive chronic pancreatitis. Pancreas 7: 193-196. [Crossref]
- Wilson JS, Colley PW, Sosula L, Pirola RC, Chapman BA, et al. (1982) Alcohol causes a fatty pancreas. A rat model of ethanol-induced pancreatic steatosis. Alcohol Clin Exp Res 6: 117-121. [Crossref]
- López JM, Bombi JA, Valderrama R, Giménez A, Parés A, Caballería J, et al. (1996) Effects of prolonged ethanol intake and malnutrition on rat pancreas. Gut 38: 285–292. [Crossref]
- Chowdhury P, Gupta P (2006) Pathophysiology of alcoholic pancreatitis: an overview. World J Gastroenterol 12: 7421-7427. [Crossref]
- Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, et al. (2013) Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 57: 601–609. [Crossref]
- Nair S, Cope K, Risby TH, Diehl AM, Terence RH (2001) Obesity and female gender increase breath ethanol concentration: potential implications for the pathogenesis of nonalcoholic steatohepatitis. Am J Gastroenterol 96: 1200–1204. [Crossref]
- Baker SS, Baker RD, Liu W, Nowak NJ, Zhu L (2010) Role of alcohol metabolism in non-alcoholic steatohepatitis. PLoS One 5: e9570. [Crossref]
- Zhu R, Baker SS, Moylan CA, Abdelmalek MF, Guy CD, et al. (2015) Systematic Analysis of NAFLD Transcriptome Revealed Elevated Expression of Alcohol Metabolizing Genes in Mild and Severe NAFLD Livers. J Pathol. 238: 531-542. [Crossref]
- Zhang Y, Baker SS, Baker RD, Zhu R, Zhu L (2012) Systematic analysis of the gene expression in the livers of nonalcoholic steatohepatitis: implications on potential biomarkers and molecular pathological mechanism. PLoS One 7: e51131. [Crossref]
- Volynets V, Küper MA, Strahl S, Maier IB, Spruss A, Wagnerberger S, et al. (2012) Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig Dis Sci 57: 1932–1941. [Crossref]
- Menezes CN, Raal F, Immelman A, Song E (2008) The role of increased gastrointestinal alcohol production in patients with the metabolic syndrome: Implications for the pathogenesis of non-alcoholic fatty liver disease. J Endocrinol Metab Diabetes South Africa 13: 48–56.
- Della Corte C, Mosca A, Majo F, Lucidi V, Panera N, Giglioni E, et al. (2015) Non-alcoholic fatty pancreas disease and Non-alcoholic fatty liver disease: more than ectopic fat. Clin Endocrinol (Oxf) 83: 656–662. [Crossref]
- Lee JS, Kim SH, Jun DW, Han JH, Jang EC, et al. (2009) Clinical implications of fatty pancreas: correlations between fatty pancreas and metabolic syndrome. World J Gastroenterol 15: 1869-1875. [Crossref]
- Uygun A, Kadayifci A, Demirci H, Saglam M, Sakin YS, et al. (2015) The effect of fatty pancreas on serum glucose parameters in patients with nonalcoholic steatohepatitis. Eur J Intern Med 26: 37-41. [Crossref]
- Herreros-Villanueva M, Hijona E, Bañales JM, Cosme A, Bujanda L (2013) Alcohol consumption on pancreatic diseases. World J Gastroenterol 19: 638-647. [Crossref]
- Karahanian E, Quintanilla ME, Tampier L, Rivera-Meza M, Bustamante D, et al. (2011) Ethanol as a prodrug: brain metabolism of ethanol mediates its reinforcing effects. Alcohol Clin Exp Res 35: 606-612. [Crossref]
- Kovach SJ, Sitzmann J V, McKillop IH (2001) Inhibition of alcohol dehydrogenase blocks enhanced Gi-protein expression following ethanol treatment in experimental hepatocellular carcinoma in vitro. Eur J Gastroenterol Hepatol 13: 1209–1216. [Crossref]
- Swaminathan K, Clemens DL, Dey A (2013) Inhibition of CYP2E1 leads to decreased malondialdehyde-acetaldehyde adduct formation in VL-17A cells under chronic alcohol exposure. Life Sci 92: 325–336. [Crossref]
- Ye Q, Wang X, Wang Q, Xia M, Zhu Y, Lian F, et al. (2013) Cytochrome P4502E1 inhibitor, chlormethiazole, decreases lipopolysaccharide-induced inflammation in rat Kupffer cells with ethanol treatment. Hepatol Res 43: 1115–1123.
- Lindros KO, Stowell L, Väänänen H, Sipponen P, Lamminsivu U, Pikkarainen P, et al. (1983) Uninterrupted prolonged ethanol oxidation as a main pathogenetic factor of alcoholic liver damage: evidence from a new liquid diet animal model. Liver 3: 79–91. [Crossref]
2021 Copyright OAT. All rights reserv
- Schaffert CS, Duryee MJ, Bennett RG, DeVeney AL, Tuma DJ, et al. (2010) Exposure of precision-cut rat liver slices to ethanol accelerates fibrogenesis. Am J Physiol Gastrointest Liver Physiol 299: G661-668. [Crossref]
- Huang W, Booth DM, Cane MC, Chvanov M, Javed MA, Elliott VL, et al. (2014) Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 63: 1313–1324. [Crossref]
- Wolin MJ, Yerry S, Miller TL, Zhang Y, Bank S (1998) Changes in production of ethanol, acids and H2 from glucose by the fecal flora of a 16- to 158-d-old breast-fed infant. J Nutr 128: 85-90. [Crossref]
- Sarkola T, Eriksson CJ (2001) Effect of 4-methylpyrazole on endogenous plasma ethanol and methanol levels in humans. Alcohol Clin Exp Res 25: 513-516. [Crossref]
- Sato H, Shiogama Y (2009) Fecal excretion of alcohols and organic anions in neonatal dairy calves. Anim Sci J 80: 171-175. [Crossref]
- Krebs HA, Perkins JR (1970) The physiological role of liver alcohol dehydrogenase. Biochem J 118: 635-644. [Crossref]
- Kapil S, Duseja A, Sharma BK, Singla B, Chakraborti A, Das A, et al. (2016) Small intestinal bacterial overgrowth and toll like receptor signaling in patients with nonalcoholic fatty liver disease. J Gastroenterol Hepatol. 31: 213-221. [Crossref]
- Ferolla SM, Armiliato GN2, Couto CA3, Ferrari TC4 (2014) The role of intestinal bacteria overgrowth in obesity-related nonalcoholic fatty liver disease. Nutrients 6: 5583-5599. [Crossref]
- Shanab AA, Scully P, Crosbie O, Buckley M, O’Mahony L, Shanahan F, et al.(2011) Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig Dis Sci 56: 1524–1534. [Crossref]
- Abu-Shanab A, Quigley EM (2010) The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 7: 691-701. [Crossref]
- Ilan Y (2012) Leaky gut and the liver: a role for bacterial translocation in nonalcoholic steatohepatitis. World J Gastroenterol 18: 2609–2618. [Crossref]
- Sabate JM, Jouet P, Harnois F, Mechler C, Msika S, Grossin M, et al. (2008) High prevalence of small intestinal bacterial overgrowth in patients with morbid obesity: a contributor to severe hepatic steatosis. Obes Surg 18: 371–377.
- Cope K, Risby T, Diehl AM. (2000) Increased gastrointestinal ethanol production in obese mice: implications for fatty liver disease pathogenesis. Gastroenterology 119: 1340–1347. [Crossref]
- Solga SF, Alkhuraishe A, Cope K, Tabesh A, Clark JM, et al. (2006) Breath biomarkers and non-alcoholic fatty liver disease: preliminary observations. Biomarkers 11: 174-183. [Crossref]
- Vakevainen S, Mentula S, Nuutinen H, Salmela KS, Jousimies-Somer H, Farkkila M, et al. (2002) Ethanol-derived microbial production of carcinogenic acetaldehyde in achlorhydric atrophic gastritis. Scand J Gastroenterol 37: 648–655. [Crossref]
- Naylor G, Axon A (2003) The role of bacterial overgrowth in the stomach as an additional risk factor for gastritis. Can J Gastroenterol. 17 Suppl B: 13B-17B [Crossref]
- Tanaka M, Fukui M, Kuroda M, Yamazaki M, Hasegawa G, et al. (2012) Pepsinogen I/II ratio is related to glucose, triacylglycerol, and uric acid levels. Nutrition 28: 418-421. [Crossref]
- Kutsuma A, Oshida H2, Suwa K3, Nakajima K4 (2014) A possible association of low pepsinogen I and pepsinogen I/II with low and high body weight in Japanese men. Clin Biochem 47: 126-128. [Crossref]
- Song H, Held M, Sandin S, Rautelin H, Eliasson M, Söderberg S, et al. (2015) Increase in the Prevalence of Atrophic Gastritis Among Adults Age 35 to 44 Years Old in Northern Sweden Between 1990 and 2009. Clin Gastroenterol Hepatol 13: 1592–600.e1.
- Kim HJ, Kim N, Kim HY, Lee HS, Yoon H, Shin CM, et al. (2015) Relationship between body mass index and the risk of early gastric cancer and dysplasia regardless of Helicobacter pylori infection. Gastric Cancer 18: 762–773. [Crossref]
- Picot D, Lauvin R, Hellegouarc’h R (1997) [Intra-digestive fermentation in intestinal malabsorption syndromes: relations with elevated serum activity of gamma-glutamyl-transpeptidase]. Gastroenterol Clin Biol 21: 562–566. [Crossref]
- Halsted CH, Robles EA, Mezey E (1973) Distribution of ethanol in the human gastrointestinal tract. Am J Clin Nutr 26: 831-834. [Crossref]
- Klipstein FA, Holdeman L V, Corcino JJ, Moore WE (1973) Enterotoxigenic intestinal bacteria in tropical sprue. Ann Intern Med 79: 632–641. [Crossref]
- Yang SL, Xia JH, Zhang YY, Fan JG, Wang H, Yuan J, et al. (2015) Hyperinsulinemia shifted energy supply from glucose to ketone bodies in early nonalcoholic steatohepatitis from high-fat high-sucrose diet induced Bama minipigs. Sci Rep 5: 13980.
- Aljomah G, Baker SS, Liu W, Kozielski R, Oluwole J, et al. (2015) Induction of CYP2E1 in non-alcoholic fatty liver diseases. Exp Mol Pathol 99: 677-681. [Crossref]
- Song BJ, Akbar M, Jo I, Hardwick JP, Abdelmegeed MA (2015) Translational Implications of the Alcohol-Metabolizing Enzymes, Including Cytochrome P450-2E1, in Alcoholic and Nonalcoholic Liver Disease. Adv Pharmacol 74: 303–372. [Crossref]
- Zhu R, Baker SS, Moylan CA, Abdelmalek MF, et al. (2016) Systematic transcriptome analysis reveals elevated expression of alcohol-metabolizing genes in NAFLD livers. J Pathol 238: 531-542. [Crossref]
- Lee SL, Chau GY, Yao CT, Wu CW, Yin SJ (2006) Functional assessment of human alcohol dehydrogenase family in ethanol metabolism: significance of first-pass metabolism. Alcohol Clin Exp Res 30: 1132–1142. [Crossref]
- Baraona E, Julkunen R, Tannenbaum L, Lieber CS (1986) Role of intestinal bacterial overgrowth in ethanol production and metabolism in rats. Gastroenterology 90: 103-110. [Crossref]
- Spinucci G, Guidetti M, Lanzoni E, Pironi L. (2006) Endogenous ethanol production in a patient with chronic intestinal pseudo-obstruction and small intestinal bacterial overgrowth. Eur J Gastroenterol Hepatol 18: 799–802. [Crossref]
- Jansson-Nettelbladt E, Meurling S, Petrini B, Sjölin J (2006) Endogenous ethanol fermentation in a child with short bowel syndrome. Acta Paediatr 95: 502-504. [Crossref]
- Bode JC, Rust S, Bode C (1984) The effect of cimetidine treatment on ethanol formation in the human stomach. Scand J Gastroenterol 19: 853-856. [Crossref]
- Borriello SP, Reed PJ, Dolby JM, Barclay FE, Webster AD (1985) Microbial and metabolic profile of achlorhydric stomach: comparison of pernicious anaemia and hypogammaglobulinaemia. J Clin Pathol 38: 946-953. [Crossref]
- Madrid AM, Hurtado C, Gatica S, Chacon I, Toyos A, Defilippi C (2002) [Endogenous ethanol production in patients with liver cirrhosis, motor alteration and bacterial overgrowth]. Rev Med Chil 130: 1329–1334. [Crossref]
- Dahshan A, Donovan K (2001) Auto-brewery syndrome in a child with short gut syndrome: case report and review of the literature. J Pediatr Gastroenterol Nutr 33: 214-215. [Crossref]
- Michail S, Lin M, Frey MR, Fanter R, Paliy O, et al. (2015) Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol Ecol 91: 1-9. [Crossref]
- Sajjad a., Mottershead M, Syn WK, Jones R, Smith S, Nwokolo CU (2005) Ciprofloxacin suppresses bacterial overgrowth, increases fasting insulin but does not correct low acylated ghrelin concentration in non-alcoholic steatohepatitis. Aliment Pharmacol Ther 22: 291–299. [Crossref]
- Elshaghabee FMF, Bockelmann W, Meske D, de Vrese M, Walte H-G, Schrezenmeir J, et al. (2016) Ethanol Production by Selected Intestinal Microorganisms and Lactic Acid Bacteria Growing under Different Nutritional Conditions. Front Microbiol 7: 47. [Crossref]
- Väkeväinen S, Mentula S, Nuutinen H, Salmela KS, Jousimies-Somer H, et al. (2002) Ethanol-derived microbial production of carcinogenic acetaldehyde in achlorhydric atrophic gastritis. Scand J Gastroenterol 37: 648-655. [Crossref]
- Roine RP, Salmela KS, Hook-Nikanne J, Kosunen TU, Salaspuro M (1992) Alcohol dehydrogenase mediated acetaldehyde production by Helicobacter pylori--a possible mechanism behind gastric injury. Life Sci 51:1333–1337. [Crossref]
- Vakevainen S, Tillonen J, Salaspuro M, Jousimies-Somer H, Nuutinen H, Farkkila M (2000) Hypochlorhydria induced by a proton pump inhibitor leads to intragastric microbial production of acetaldehyde from ethanol. Aliment Pharmacol Ther 14: 1511–1518. [Crossref]
- Väkeväinen S, Tillonen J, Blom M, Jousimies-Somer H, Salaspuro M (2001) Acetaldehyde production and other ADH-related characteristics of aerobic bacteria isolated from hypochlorhydric human stomach. Alcohol Clin Exp Res 25: 421–426. [Crossref]
- Salmela KS, Roine RP, Koivisto T, Höök-Nikanne J, Kosunen TU, et al. (1993) Characteristics of Helicobacter pylori alcohol dehydrogenase. Gastroenterology 105: 325-330. [Crossref]
- Tsuruya A, Kuwahara A, Saito Y, Yamaguchi H, Tenma N, Inai M, et al. (2016) Major Anaerobic Bacteria Responsible for the Production of Carcinogenic Acetaldehyde from Ethanol in the Colon and Rectum. Alcohol Alcohol pii: agv135.
- Jokelainen K, Roine RP, Väänänen H, Färkkilä M, Salaspuro M (1994) In vitro acetaldehyde formation by human colonic bacteria. Gut 35: 1271-1274. [Crossref]
- Jokelainen K, Matysiak-Budnik T, Mäkisalo H, Höckerstedt K, Salaspuro M (1996) High intracolonic acetaldehyde values produced by a bacteriocolonic pathway for ethanol oxidation in piglets. Gut 39: 100–104. [Crossref]
- Tillonen J, Kaihovaara P, Jousimies-Somer H, Heine R, Salaspuro M (1998) Role of catalase in in vitro acetaldehyde formation by human colonic contents. Alcohol Clin Exp Res 22: 1113–1119. [Crossref]
- Hiele M, Ghoos Y, Rutgeerts P, Vantrappen G, Schoorens D (1991) Influence of nutritional substrates on the formation of volatiles by the fecal flora. Gastroenterology 100: 1597-1602. [Crossref]
- Kurkivuori J, Salaspuro V, Kaihovaara P, Kari K, Rautemaa R, et al. (2007) Acetaldehyde production from ethanol by oral streptococci. Oral Oncol 43: 181-186. [Crossref]
- Jokelainen K, Roine RP, Väänänen H, Färkkilä M, Salaspuro M (1994) In vitro acetaldehyde formation by human colonic bacteria. Gut 35: 1271-1274. [Crossref]
- Pronko P, Bardina L, Satanovskaya V, Kuzmich A, Zimatkin S (2002) Effect of chronic alcohol consumption on the ethanol- and acetaldehyde-metabolizing systems in the rat gastrointestinal tract. Alcohol Alcohol 37: 229–235. [Crossref]
- Nuutinen H, Lindros KO, Salaspuro M (1983) Determinants of blood acetaldehyde level during ethanol oxidation in chronic alcoholics. Alcohol Clin Exp Res 7: 163–168. [Crossref]
- Mello T, Ceni E, Surrenti C, Galli A (2008) Alcohol induced hepatic fibrosis: role of acetaldehyde. Mol Aspects Med 29: 17-21. [Crossref]
- Moon KH, Abdelmegeed MA, Song BJ (2007) Inactivation of cytosolic aldehyde dehydrogenase via S-nitrosylation in ethanol-exposed rat liver. FEBS Lett 581: 3967-3972. [Crossref]
- Alderman JA, Sanny C, Gordon E, Lieber CS (1985) Ethanol feeding can produce secondary alterations in aldehyde dehydrogenase isozymes. Alcohol 2: 91-95. [Crossref]
- Thomas M, Halsall S, Peters TJ (1982) Role of hepatic acetaldehyde dehydrogenase in alcoholism: demonstration of persistent reduction of cytosolic activity in abstaining patients. Lancet 2: 1057–1058. [Crossref]
- Palmer KR, Jenkins WJ (1982) Impaired acetaldehyde oxidation in alcoholics. Gut 23: 729-733. [Crossref]
- Lands WE (1998) A review of alcohol clearance in humans. Alcohol 15: 147–160. [Crossref]
- Logan BK, Jones AW (2000) Endogenous ethanol 'auto-brewery syndrome' as a drunk-driving defence challenge. Med Sci Law 40: 206-215. [Crossref]
- Kaji H, Asanuma Y, Yahara O, Shibue H, Hisamura M, Saito N, et al. (1984) Intragastrointestinal alcohol fermentation syndrome: report of two cases and review of the literature. J Forensic Sci Soc 24: 461–471. [Crossref]
- Logan BK, Jones AW (2003) Endogenous ethanol production in a child with short gut syndrome. J Pediatr Gastroenterol Nutr 36: 419-420. [Crossref]
- Caprara DL, Klein J, Koren G (2005) Baseline measures of fatty acid ethyl esters in hair of neonates born to abstaining or mild social drinking mothers. Ther Drug Monit 27: 811–815. [Crossref]
- Chan D, Bar-Oz B, Pellerin B, Paciorek C, Klein J, et al. (2003) Population baseline of meconium fatty acid ethyl esters among infants of nondrinking women in Jerusalem and Toronto. Ther Drug Monit 25: 271-278. [Crossref]
- Zhu L, Baker RD, Baker SS (2015) Gut microbiome and nonalcoholic fatty liver diseases. Pediatr Res 77: 245-251. [Crossref]
- Tomita K, Teratani T, Yokoyama H, Suzuki T, Irie R, et al. (2011) Plasma free myristic acid proportion is a predictor of nonalcoholic steatohepatitis. Dig Dis Sci 56: 3045-3052. [Crossref]
- Sundaresan S, Vijayagopal P, Mills N, Imrhan V, Prasad C (2011) A mouse model for nonalcoholic steatohepatitis. J Nutr Biochem 22: 979-984. [Crossref]
- Lee JJ, Lambert JE, Hovhannisyan Y, Ramos-Roman MA, Trombold JR, Wagner DA, et al. (2015) Palmitoleic acid is elevated in fatty liver disease and reflects hepatic lipogenesis. Am J Clin Nutr 101: 34–43. [Crossref]
- Lê KA, Ventura EE, Fisher JQ, Davis JN, Weigensberg MJ, et al. (2011) Ethnic differences in pancreatic fat accumulation and its relationship with other fat depots and inflammatory markers. Diabetes Care 34: 485-490. [Crossref]
- Chiang CP, Wu CW, Lee SP, Chung CC, Wang CW, Lee SL, et al. (2009) Expression pattern, ethanol-metabolizing activities, and cellular localization of alcohol and aldehyde dehydrogenases in human pancreas: implications for pathogenesis of alcohol-induced pancreatic injury. Alcohol Clin Exp Res 33: 1059–1068. [Crossref]
- He ZJ, Ericksson P, Alho H, Harmoinen A, Nordback I (2001) Effects on endogenous acetaldehyde production by disulfiram and ethanol feeding on rat pancreas. J Gastrointest Surg 5: 531-539. [Crossref]
- Honda S, Fujioka T, Shiota K, Fujiyama K, Kubota T, et al. (1995) Effects of ethanol on the pancreas of disulfiram-treated rats. J Gastroenterol 30: 231-236. [Crossref]
- Apte M, McCarroll J, Pirola R, Wilson J (2007) Pancreatic MAP kinase pathways and acetaldehyde. Novartis Found Symp 285: 200-211. [Crossref]
- Shimizu K (2008) Mechanisms of pancreatic fibrosis and applications to the treatment of chronic pancreatitis. J Gastroenterol 43: 823-832. [Crossref]
- Vonlaufen A, Wilson JS, Pirola RC, Apte MV (2007) Role of alcohol metabolism in chronic pancreatitis. Alcohol Res Health 30: 48-54. [Crossref]
- Crous-Bou M, Porta M, López T, Jariod M, Malats N, Morales E, et al. (2009) Lifetime history of alcohol consumption and K-ras mutations in pancreatic ductal adenocarcinoma. Environ Mol Mutagen 50: 421–430. [Crossref]
- Singh S, Arcaroli J, Thompson DC, Messersmith W, Vasiliou V (2015) Acetaldehyde and retinaldehyde-metabolizing enzymes in colon and pancreatic cancers. Adv Exp Med Biol 815: 281-294. [Crossref]
- Matysiak-Budnik T, Jokelainen K, Kärkkäinen P, Mäkisalo H, Ohisalo J, et al. (1996) Hepatotoxicity and absorption of extrahepatic acetaldehyde in rats. J Pathol 178: 469-474. [Crossref]
- BaÅkowski E, Pawlicka E, Sobolewski K (1993) Liver collagen of rats submitted to chronic intoxication with acetaldehyde. Mol Cell Biochem 121: 37-43. [Crossref]
- Criddle DN (2015) The role of fat and alcohol in acute pancreatitis: A dangerous liaison. Pancreatology 15: S6-6S12. [Crossref]
- Werner J, Saghir M, Warshaw AL, Lewandrowski KB, Laposata M, et al. (2002) Alcoholic pancreatitis in rats: injury from nonoxidative metabolites of ethanol. Am J Physiol Gastrointest Liver Physiol 283: G65-73. [Crossref]
- Werner J, Laposata M, Fernández-del Castillo C, Saghir M, Iozzo RV, et al. (1997) Pancreatic injury in rats induced by fatty acid ethyl ester, a nonoxidative metabolite of alcohol. Gastroenterology 113: 286-294. [Crossref]
- Lugea A, Gukovsky I, Gukovskaya AS, Pandol SJ (2003) Nonoxidative ethanol metabolites alter extracellular matrix protein content in rat pancreas. Gastroenterology 125: 1845–1859. [Crossref]
- Brick J (2006) Standardization of alcohol calculations in research. Alcohol Clin Exp Res 30: 1276-1287. [Crossref]
- Engstler AJ, Aumiller T, Degen C, Dürr M, Weiss E, Maier IB, et al. (2015) Insulin resistance alters hepatic ethanol metabolism: studies in mice and children with non-alcoholic fatty liver disease. Gut pii: gutjnl-2014-308379. [Crossref]
- Lee SL, Wang MF, Lee AI, Yin SJ (2003) The metabolic role of human ADH3 functioning as ethanol dehydrogenase. FEBS Lett 544: 143-147. [Crossref]
- Masters SB, Trevor AJ (2015) The Alcohols. In: Katzung BG, Trevor AJ, editors. Basic Clin. Pharmacol. 13e, New York, McGraw-Hill Medical.
- Ropper AH, Samuels MA, Klein JP (2014) Chapter 42. Alcohol and Alcoholism. Adams Victor’s Princ. Neurol. 10e, New York, The McGraw-Hill Companies.
- Mezey E, Imbembo AL, Potter JJ, Rent KC, Lombardo R, Holt PR (1975) Endogenous ethanol production and hepatic disease following jejunoileal bypass for morbid obesity. Am J Clin Nutr 28: 1277–1283.
- Bergheim I, Weber S, Vos M, Krämer S, Volynets V, et al. (2008) Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: role of endotoxin. J Hepatol 48: 983-992. [Crossref]
- Di Luccia B, Crescenzo R, Mazzoli A, Cigliano L, Venditti P, et al. (2015) Rescue of Fructose-Induced Metabolic Syndrome by Antibiotics or Faecal Transplantation in a Rat Model of Obesity. PLoS One 10: e0134893. [Crossref]
- Membrez M, Blancher F, Jaquet M, Bibiloni R, Cani PD, et al. (2008) Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J 22: 2416-2426. [Crossref]
- Lichtman SN, Keku J, Schwab JH, Sartor RB (1991) Hepatic injury associated with small bowel bacterial overgrowth in rats is prevented by metronidazole and tetracycline. Gastroenterology 100: 513-519. [Crossref]
- Vanderhoof JA, Tuma DJ, Antonson DL, Sorrell MF (1982) Effect of antibiotics in the prevention of jejunoileal bypass-induced liver dysfunction. Digestion 23: 9-15. [Crossref]
- Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG (1995) Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 108: 218–224. [Crossref]
- Rabot S, Membrez M, Bruneau A, Gérard P, Harach T, et al. (2010) Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J 24: 4948-4959. [Crossref]
- Frazier TH, DiBaise JK, McClain CJ (2011) Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. JPEN J Parenter Enteral Nutr 35: 14S-20S. [Crossref]
- Caesar R, Reigstad CS, Backhed HK, Reinhardt C, Ketonen M, Ostergren Lunden G, et al. (2012) Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut 61:1701–1707.
- M C C C, N L L, C M F, J L G, D A, et al. (2014) Comparing the effects of acute alcohol consumption in germ-free and conventional mice: the role of the gut microbiota. BMC Microbiol 14: 240. [Crossref]
- Cederbaum AI (2012) Alcohol metabolism. Clin Liver Dis 16: 667-685. [Crossref]
- Jadhav SB, Bankar SB, Granström T, Ojamo H, Singhal RS, et al. (2015) Interaction of carbohydrates with alcohol dehydrogenase: Effect on enzyme activity. J Biosci Bioeng 120: 252-256. [Crossref]
- Haseba T, Ohno Y (2010) A new view of alcohol metabolism and alcoholism--role of the high-Km Class III alcohol dehydrogenase (ADH3). Int J Environ Res Public Health 7: 1076-1092. [Crossref]
- Yin SJ, Chou CF, Lai CL, Lee SL, Han CL (2013) Human class IV alcohol dehydrogenase: kinetic mechanism, functional roles and medical relevance. Chem Biol Interact 143-144: 219–227. [Crossref]
- Levitt MD, Levitt DG (2000) Appropriate use and misuse of blood concentration measurements to quantitate first-pass metabolism. J Lab Clin Med 136: 275-280. [Crossref]
- Jones AW, Neri A (1991) Evaluation of Blood-Ethanol Profiles after Consumption of Alcohol Together with a Large Meal. Can Soc Forensic Sci J 24: 165–173.
- Paton A (2005) Alcohol in the body. BMJ 330: 85-87. [Crossref]
- Nakajima S, Hira T, Hara H (2015) Postprandial glucagon-like peptide-1 secretion is increased during the progression of glucose intolerance and obesity in high-fat/high-sucrose diet-fed rats. Br J Nutr 113: 1477-1488. [Crossref]
- Soza A, Riquelme A, González R, Alvarez M, Pérez-Ayuso RM, Glasinovic JC, et al. (2005) Increased orocecal transit time in patients with nonalcoholic fatty liver disease. Dig Dis Sci 50:1136–1140. [Crossref]
- Kinoshita H, Ijiri I, Ameno S, Fuke C, Fujisawa Y, Ameno K (1996) Inhibitory mechanism of intestinal ethanol absorption induced by high acetaldehyde concentrations: effect of intestinal blood flow and substance specificity. Alcohol Clin Exp Res 20: 510–513. [Crossref]
- Shinohara T, Ijiri I, Ameno S, Fuke C, Ameno K (1993) A comparative study of ethanol absorption in the canine jejunum after pretreatment with cyanamide or pyrazole. Alcohol Alcohol 28: 423-429. [Crossref]
- Werner J, Saghir M, Fernandez-del Castillo C, Warshaw AL, Laposata M (2001) Linkage of oxidative and nonoxidative ethanol metabolism in the pancreas and toxicity of nonoxidative ethanol metabolites for pancreatic acinar cells. Surgery 129: 736–744. [Crossref]
- Pfützer RH, Tadic SD, Li H-S, Thompson BS, Zhang J-Y, Ford ME, et al. (2002) Pancreatic cholesterol esterase, ES-10, and fatty acid ethyl ester synthase III gene expression are increased in the pancreas and liver but not in the brain or heart with long-term ethanol feeding in rats. Pancreas 25: 101–106. [Crossref]
- Criddle DN, Sutton R, Petersen OH (2006) Role of Ca2+ in pancreatic cell death induced by alcohol metabolites. J Gastroenterol Hepatol 21 Suppl 3: S14-17. [Crossref]
- Fjeld K, Weiss FU, Lasher D, Rosendahl J, Chen JM, et al. (2015) A recombined allele of the lipase gene CEL and its pseudogene CELP confers susceptibility to chronic pancreatitis. Nat Genet 47: 518-522. [Crossref]
- Whitcomb DC (2013) Genetic risk factors for pancreatic disorders. Gastroenterology 144: 1292-1302. [Crossref]
- Weiss FU, Schurmann C, Guenther A, Ernst F, Teumer A, Mayerle J, et al. (2015) Fucosyltransferase 2 (FUT2) non-secretor status and blood group B are associated with elevated serum lipase activity in asymptomatic subjects, and an increased risk for chronic pancreatitis: a genetic association study. Gut 64: 646–656. [Crossref]
- Miyasaka K, Ohta M, Takano S, Hayashi H, Higuchi S, et al. (2005) Carboxylester lipase gene polymorphism as a risk of alcohol-induced pancreatitis. Pancreas 30: e87-91. [Crossref]
- Zhong Y, Cao J, Zou R, Peng M (2015) Genetic polymorphisms in alcohol dehydrogenase, aldehyde dehydrogenase and alcoholic chronic pancreatitis susceptibility: a meta-analysis. Gastroenterol Hepatol 38: 417–425.
- Rebours V, Gaujoux S, d’Assignies G, Sauvanet A, Ruszniewski P, Levy P, et al. (2015) Obesity and Fatty Pancreatic Infiltration Are Risk Factors for Pancreatic Precancerous Lesions (PanIN). Clin Cancer Res 21: 3522–3528.
- Dobbins M, Decorby K, Choi BC (2013) The Association between Obesity and Cancer Risk: A Meta-Analysis of Observational Studies from 1985 to 2011. ISRN Prev Med 680536. [Crossref]
- Burke CA (2010) Colonic complications of obesity. Gastroenterol Clin North Am 39: 47–55. [Crossref]
- Charrez B, Qiao L, Hebbard L (2015) The role of fructose in metabolism and cancer. Horm Mol Biol Clin Investig 22: 79-89. [Crossref]
- Ungefroren H, Gieseler F, Fliedner S, Lehnert H (2015) Obesity and cancer. Horm Mol Biol Clin Investig 21: 5-15. [Crossref]
- Montella M, Di Maso M, Crispo A, Grimaldi M, Bosetti C, et al. (2015) Metabolic syndrome and the risk of urothelial carcinoma of the bladder: a case-control study. BMC Cancer 15: 720. [Crossref]
- Kent BD, McNicholas WT, Ryan S (2015) Insulin resistance, glucose intolerance and diabetes mellitus in obstructive sleep apnoea. J Thorac Dis 7: 1343-1357. [Crossref]
- Briançon-Marjollet A, Weiszenstein M, Henri M, Thomas A, Godin-Ribuot D, Polak J. (2015) The impact of sleep disorders on glucose metabolism: endocrine and molecular mechanisms. Diabetol Metab Syndr 7: 25. [Crossref]
- Matsushita N, Osaka T, Haruta I, et al. (2016) Effect of Lipopolysaccharide on the Progression of Non-Alcoholic Fatty Liver Disease in High Caloric Diet-Fed Mice. Scand J Immunol 83: 109-118. [Crossref]
- Ceccarelli S, Panera N, Mina M, Gnani D, De Stefanis C, Crudele A, et al. (2015) LPS-induced TNF-a factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget 6: 41434–41452.
- Guo H, Diao N, Yuan R, Chen K, Geng S, et al. (2016) Subclinical-Dose Endotoxin Sustains Low-Grade Inflammation and Exacerbates Steatohepatitis in High-Fat Diet-Fed Mice. J Immunol 196: 2300-2308. [Crossref]
- Murata T1, Miwa I, Toyoda Y, Okuda J (1993) Inhibition of glucose-induced insulin secretion through inactivation of glucokinase by glyceraldehyde. Diabetes 42: 1003-1009. [Crossref]
- Matafome P, Sena C, Seiça R (2013) Methylglyoxal, obesity, and diabetes. Endocrine 43: 472-484. [Crossref]
- Fiory F, Lombardi A, Miele C, Giudicelli J, Beguinot F, Van Obberghen E (2011) Methylglyoxal impairs insulin signalling and insulin action on glucose-induced insulin secretion in the pancreatic beta cell line INS-1E. Diabetologia 54: 2941–2952.
- Vander Jagt DL (2008) Methylglyoxal, diabetes mellitus and diabetic complications. Drug Metabol Drug Interact 23: 93-124. [Crossref]
- Hachiya H, Miura Y, Inoue K-I, Park KH, Takeuchi M, Kubota K. (2014) Advanced glycation end products impair glucose-induced insulin secretion from rat pancreatic ß-cells. J Hepatobiliary Pancreat Sci 21: 134–141.
- Zhu Y, Shu T, Lin Y, Wang H, Yang J, et al. (2011) Inhibition of the receptor for advanced glycation endproducts (RAGE) protects pancreatic β-cells. Biochem Biophys Res Commun 404: 159-165. [Crossref]
- Wiseman DA, Thurmond DC (2012) The good and bad effects of cysteine S-nitrosylation and tyrosine nitration upon insulin exocytosis: a balancing act. Curr Diabetes Rev 8: 303-315. [Crossref]
- Mattson MP (2009) Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol 44: 625-633. [Crossref]
- Marnett LJ (2002) Oxy radicals, lipid peroxidation and DNA damage. Toxicology 181-182: 219-222. [Crossref]