Raxatrigine (5R)-5-{4-[(2-fluorobenzyl)oxy]phenyl}-L-prolinamide; chemical formula: C18H19FN2O2) is a sodium channel blocker first synthesized by GlaxoSmithKline, explored and developed under de code name GSK1014802 as a central NaV1.3 blocker up to and including phase I in the period 2005-2009. Related to its central mechanism of action, the target indication selected at that time was bipolar disorder, pain and perhaps epilepsy. Subsequently the Glaxo group revamped its R&D focus and stopped working in the field of pain around 2010. The Glaxo research group working on sodium channels left the company and created a spin-out: Covergence Pharmaceuticals, taking with them the rights on the compound, renamed CNV1014802. The characterization of the compound changed into a sodium channel inhibitor reported to have high selectivity for the peripheral Nav1.7 subtype channel, and phase II development started in neuropathic pain, in lumbosacral radiculopathy and trigeminal neuralgia. In 2015 Convergence was taken over by Biogen and the compound was recoded BIB074. Recent published data do not characterize the compound as a selective Nav1.7 inhibitor, but rather as an unspecific sodium channel blocker. We will analyze scientific and press communications referring to this compound in the period 2005-2016 and discuss the moving target for the compound.
GSK1014802, raxatrigine, CNV1014802, sodium channels, NaV1.7, NaV1.3, neuropathic pain
Currently raxatrigine is in phase II/III for lumbosacral radiculopathy and in phase IIa for erythromelalgia and is developed by the Biogen subsidiary Convergence. There is not much published scientific literature on the compound. In PubMed we find only 3 articles using the key word ‘CNV1014802’. The oldest article written by amongst others company representatives. It covers the design of a phase II study, while the article also offers a review of the preclinical profile of the drug, however without any references to primary sources [1]. A second article is a review and discuss only some generalities related to the drug [2]. The most recent article is from independent researchers from the Universities of Queensland (AU), Sheffield (UK) and Erlangen(D). The compound in their hands is an aspecific sodium channel blocker, with affinity for nearly all channels in the same order of magnitude [3]. There is only one article when searching on ‘GSK1014802’ related to bipolar disorders and it does not discuss any primary data [4].
Further sources of information are a number of patents and two more abstracts are available presenting phase II data, as well as a number of company presentations and press releases on the web.
In 2006 the Glaxo group filed a patent: prolinamide derivatives as sodium channel modulators (patent WO 2007042239 A1, priority date 10 October 2005). In this patent the compound 5-(4-{[(2-Fluorophenyl)methyl]oxy}phenyl)-prolinamide was patented, as well as the synthesis, and its use as a pharmaceutical composition for the treatment or prevention of disorders via the modulation of voltage gated sodium channels. Three indications were highlighted: depression, bipolar mood disorder and substance disorders. In one embodiment of the invention the compound had to be a use dependent sodium channel inhibitor and more specifically a subtype NaV1.3 sodium channel use dependent inhibitor. Nav 1.3 was defined in the patent as a brain specific sodium channel (together with NaV1.1 , 1.2, and 1.6.). Furthermore, the compound needed to have a suitable profile, amongst others in terms of exposure (Cmax) and/or bioavailability on oral administration. In the patent raxatrigine was referred to as formula (I).
The biological activity of the compound was tested in the patent in stable cell lines expressing hNaV1.3 channels, created via the transfecting of CHO cells with the pCIN5-hNav1.3 vector. Raxatrigine was tested in this assay and the pUD-15 values were greater than 5.0. No other assays were used, so based on this patent the only conclusion could be that raxatrigine was a sodium channel blocker with high affinity for the Nav1.3 channel. These channels were since 2003 known to be upregulated in central neuropathic pain states [5].
Glaxo Group continued after preclinical studies setting up a phase I program around 2008, consisting of at least 3 phase I studies and referred to the compound as a use-dependent sodium channel blocker and an effective anticonvulsant in animal models. The focus was the treatment of pain, and the third phase I study was indeed designed to evaluate surrogate parameters related to the analgesic effects of the drug. The phase I program consisted of:
- A study on the safety, tolerability and pharmacokinetics of repeated doses of the compound administered for up to 28 days in healthy male or female subjects, including a food interaction study.
- A randomized, double-blind, placebo-controlled, repeat dose study, in approximately 60 subjects to receive GSK1014802 400 mg bid and placebo for 36 days with at least 1 week between treatment sessions, including monitoring ambulatory blood pressure.
- A double blind, placebo controlled, 4-period cross over study in 20 subjects testing one of two doses of the drug or of lidocaine versus placebo with at least 2 weeks between sessions.
In 2009 Glaxo Group presented its pipeline and presented raxatrigine as compound 1014802, a sodium channel blocker for bipolar disorder in phase l [6]. In that year the transition of the indication bipolar disorder to pain must have taken place. In 2010 Glaxo Group refocused her strategic fields and wanted to exit the pain field. In that process, Convergence Pharmaceuticals was created in 2010 by 12 scientists from GlaxoSmithKline and they acquired the rights on the compound GSK1014802, renamed as CNV1014802 and a calcium channel ligand. In a press release of 27-10-2010 CNV1014802 was characterized as ‘a potent, state-dependent sodium channel blocker’ [7].
Covergence: start of CNV1014802 as a Nav1.7 sodium channel blocker
Dr. Ged Giblin, head of chemistry and preclinical development at Convergence characterized the drug subsequently in an interview as the lead neuropathic pain candidate (CNV1014802) of the company: ‘a small-molecule, state-dependent sodium channel blocker targeting the Nav1.7 sodium channel’ [8]. It was unclear why and when the compound changed from a Nav1.3 to a Nav1.7 blocker. Since that time the compound was referred to as such a blocker, for instance in a press release: ‘Convergence Pharmaceuticals Announces Start of Phase II Study for its Selective Nav1.7 blocker CNV1014802, July 19, 2011’ [9]. Interestingly, in 2014 the compound was characterized by independent scientists as: ‘a Nav1.7 channel inhibitors with unknown selectivity’ [10].
Convergence developed the compound up to and including phase II for 2 indications: lumbar radiculopathy and trigeminal pain. Raxatrigine received orphan-drug designation for the treatment of trigeminal neuralgia from the US Food and Drug Administration in July 2013. The compound is discontinued in bipolar depression [11]. Convergence was acquired by Biogen in 2015 and the compound is currently developed under de the code-name BOB074 by Convergence, which is now as a subsidiary of Biogen. Biogen on its company website characterizes the compound as a Nav1.7 inhibitor in phase II [12]. The site only refers to trigeminal neuralgia as indication in development. However, in ClinTrials.gov no further phase III studies in this indication are mentioned apart from a completed phase II study (see under), while a full powered phase II 2 dose-regimes study in radiculopathy is ongoing and recruiting (630 patients in the period 2016-2017; high dose, low dose, placebo).
The preclinical profile of raxatrigine is hidden in a clinical paper discussing the design of a clinical trial in trigeminal neuralgia.1 That is unfortunate, because therefore the profile is not easy to find, and more importantly, there are no primary sources mentioned in the paper, it is just a summary of findings, without context.
Raxatrigine is described by the authors (some authors are from Covergence) as a peripherally and centrally acting agent inhibiting sodium channels in a state-dependent fashion. The authors state the compound shows selectivity for the Nav1.7 subtype over the other subtypes tested (Nav1.1, Nav1.2, Nav1.3, Nav1.5, Nav1.6 and TTX-R), for both the resting and depolarized states. The amount of sodium channel block increases with the frequency of stimulation for Nav1.7 and for Nav1.2 and Nav1.6. The block is said to be more activity-driven at Nav1.2 and Nav1.6 than it is at Nav1.7. CNV1014802 is also said to preferentially target and inhibit higher frequencies of firing (from 10 Hz onwards) induced by noxious stimuli or during seizures. We could not find any other sources for this preclinical profile. That is unfortunate, since now we are unable to understand why a more recent paper came to quite different conclusions. Neither can we understand why in one of the first patent the compound was characterized as a Nav1.3 blocker only.
In a recent paper published by Deuis et al. CNV1014802 is characterized as a Non-Selective NaV Inhibitor. The authors start pointing out that CNV1014802 is reported to be a state-dependent inhibitor of NaV1.7, but information on the potency and selectivity profile has not been reported in literature. The authors tested the compound in a special assay and found the compound to have the following profile in humanized NaV channels: in rank order of potency (pIC50 ± SEM): NaV1.8 (5.25 ± 0.1) > NaV1.4 (5.09 ± 0.2) > NaV1.2 (4.99 ± 0.2) > NaV1.6 (4.84 ± 0.1) > NaV1.3 (4.82 ± 0.3) > NaV1.1 (4.70 ± 0.2) > NaV1.7 (4.58 ± 0.2) > NaV1.5 (4.18 ± 0.2). They also documented a state-dependency with a nine-fold shift in the NaV1.7 IC50 between closed/resting state inhibition (54 μM) and open/inactivated state inhibition (6.3 μM), with a clear preference for the open/inactivated state. They could compare this profile with that from the Pfizer sodium channel blocker PF-04856264, which indeed is a selective NaV1.7 inhibitor, and this novel aryl sulfonamide NaV1.7 inhibitor was 50 times more potent at the NaV1.7 channel. Intraplantar administration of CNV1014802 (1 mM) had no significant effect on spontaneous pain behaviors in mice evoked by the scorpion toxin OD1, but intraperitoneal administration of CNV1014802 (3 and 30 mg/kg) did reduced spontaneous pain behaviors. 30 mg/kg of PF-04856264 and 30 mg/kg CNV1014802 had comparable analgesic effects after i.p. injection.
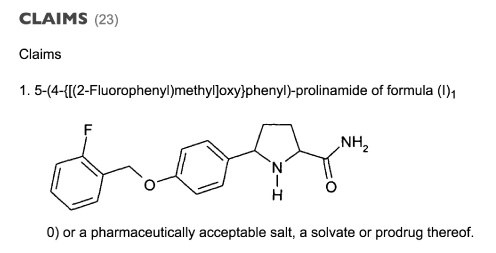
Figure 1. The first claim from the Glaxo Group patent of 2005 claims the structure as above, which is raxatrigine, and its 3 diastereoisomers, its salt, solvate or prodrug for the treatment of depression, bipolar disorders, substance abuse disorders and pain.
Drug Research and Development is a highly complex endeavor, and companies should understand that it is important to first publish data in peer reviewed journals, before scientific facts are communicated in the lay press, or in press releases or during company presentations for shareholders. Scientific company representatives should not characterize pipeline drugs based on pharmacological and clinical properties, when the primary data have not been published yet. The raxatrigine case analyzed in this article serves as an example to point out why such recommendations are vital for the success and credibility of pharma and biotech industries.
- Zakrzewska JM, Palmer J, Ettlin DA, Obermann M, Giblin GM, et al. (2013) Novel design for a phase IIa placebo-controlled, double-blind randomized withdrawal study to evaluate the safety and efficacy of CNV1014802 in patients with trigeminal neuralgia. Trials 14: 402. [Crossref]
- Bagal SK, Chapman ML, Marron BE, Prime R, Storer RI, et al. (2014) Recent progress in sodium channel modulators for pain. Bioorg Med Chem Lett 24: 3690-3699. [Crossref]
- Deuis JR, Wingerd JS, Winter Z, Durek T, Dekan Z, et al. (2016) Analgesic Effects of GpTx-1, PF-04856264 and CNV1014802 in a Mouse Model of NaV1.7-Mediated Pain. Toxins (Basel) 8. [Crossref]
- El-Mallakh RS, Elmaadawi AZ, Gao Y, Lohano K, Roberts RJ (2011) Current and emerging therapies for the management of bipolar disorders. J Cent Nerv Syst Dis 3: 189-197.
- Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, et al. (2003) Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci 23: 8881-8892. [Crossref]
- GSK Product Development Pipeline February 2009.
- Former GSK employees form independent biotechnology company,
- Companies converge on pain. Drug Discovery News, January 2011;7:1:page 3.
- http://www.nlvpartners.com/press_archive.html
- Sun S, Cohen CJ, Dehnhardt CM (2014) Inhibitors of voltage-gated sodium channel Nav1.7: patent applications since 2010. Pharm Pat Anal 3: 509-521. [Crossref]
- http://adisinsight.springer.com/drugs/800027679
- https://www.biogen.com/en_us/research-pipeline/biogen-pipeline.html