Abstract
Helicobacter pylori (H. pylori) are gram-negative spiral bacterium that is believed to be one of the main causes of chronic gastritis affecting nearly one half of world population and is a root cause of complications like mucosal atrophy, gastric carcinoma, or gastric lymphoma. The current state of the art literature describes the emergence of antibiotic resistant strains and unavoidable side effects of current therapies, therefore it becomes essential to find new, safe, effective and affordable drugs and identify new drug targets. Arginase activity is known to contribute significantly in the pathophysiology of H. pylori infections, thus making it an attractive drug target. Present investigation deals with designing the theoretical model of arginase from H. pylori followed by refinement using molecular dynamics (MD) and validation using molecular docking and paired potential analysis. Models generated using Modeller and SwissModel server were evaluated using QMEAN, ProCheck, WhatCheck/WhatIf and Verify 3D. NAMD was utilized to perform conventional MD analysis. AutoDock 4.2 and ArgusLab were employed for docking investigation and paired potential analysis was done using DrugScore potential available from online DSX-server. The proposed model qualified on geometrical, stereo-chemical and other structure validation tests possessing highly conserved active site. The MD results confirm thermodynamic and conformational stability. Docking and paired potential analysis indicate involvement of highly conserved residues in stabilization of arginase-inhibitor complexes. We propose structure of arginase, using homology modeling approach. Predicted models have passed standard evaluation tests indicating good quality models. Docking and paired-potential analysis studies revealed novel interactions that have potential to be targeted in developing further structure based drug-design experiments.
Key Words
homology modeling; molecular docking; molecular dynamics; paired potential analysis; DrugScore potential; NAMD
Introduction
Helicobacter pylori (H. pylori) are small microaerophilic, gram negative, curved, bacteria that colonize and grow around the luminal surface of the gastric epithelium. Since its discovery in 1984, plethora of literature has accumulated linking its virulent role in gastritis, peptic ulcers, duodenal ulcers and colon cancer [1,2]. Because of the persistence association of H. pylori with gastric cancer, in 1994 International Agency for Research on Cancer (IARC), has classified H. pylori infection as a group 1 carcinogenic agent [3,4]. Overall, H. pylori infections are highly attributed with poor survival and very high morbidity rate were observed in majority of the clinical studies. The first line therapy, as per European [5] and North American [6] guidelines for treating H. pylori infection, prescribes a proton pump inhibitor (PPI) or ranitidine bismuth citrate, along with any two antibiotics among amoxicillin, clarithromycin and metronidazole, to be given for 7 to 14 days to adults. Up to 50 percent of patients experience side effects while taking H. pylori treatment that includes hypergastrinaemia [7], atrophic gastritis [8] and even gastric neoplasia [9-11]. Moreover, the evolving multidrug resistant strains [12, 13] limit the usage and efficacy of the currently used therapeutic modalities for the management of H. pylori infection. The clinical, epidemiological and the circumstantial literature cited above clearly warrants the need of developing novel, effective and target oriented therapeutic approach for the management of H. pylori infection. In the mainstream of identifying novel therapeutic targets for designing effective anti H. pylori agents, arginase seems to be a potential candidate because of its relevance with the survival of H. pylori. Arginase is a metalloenzyme, catalyzing the conversion of L-arginine to L-ornithine and urea [14]. In the subsequent urease driven enzymatic reaction, urea is converted into carbon dioxide and ammonia [15]. The ammonia thus liberated is utilized by H. pylori for its survival, because H. pylori is highly sensitive to acidic environment [16,17]. Thus, inhibition of arginase activity may deplete the local supply of urea for ammonia production by urease activity, and thereby arrest the growth of H. pylori. Hence modeling and structural analysis of enzyme arginase is important for identification of potential inhibitor binding sites. Experimental techniques to solve the structure of proteins are very tedious and prolonged and not always succeed in determining structure for all proteins, especially membrane proteins [18,19], thus computational techniques like homology modeling is one of the alternative approach to obtain reliable structural models for target proteins that can be utilized in structure based drug design process. Understanding the structural aspects of arginase is important for unraveling the secrets of inhibitor binding sites, which can be further explored for the design and development of novel therapeutic agents against H. pylori infections. In the present study we have designed the theoretical model of arginase from H. pylori, India 7 strain. Molecular dynamics (MD) approach was used to refine the model followed by validating the model by docking its inhibitor using Autodock 4.2 and ArgusLab. Moreover, we have also tested docking of some experimental and structural analogs of arginine such as ornithine, which is the most potent competitive inhibitor and nor-valine as non-competitive inhibitor of arginase [20]. Some prominent inhibitors of arginase like DMFO and new inhibitor S-nitroso-N-acetylpenicillamine (SNAP) [11,21] were also analyzed by docking onto arginase model. We have also carried out paired potential analysis to identify regions in protein that are involved in favorable and unfavorable interactions with ligands using DSX online knowledge-based scoring function [22].
Methods
Sequence retrieval and template identification
Arginase sequence of H. pylori was retrieved from SwissProt [23] with accession number E8QE13. In order to find suitable template for modeling, two approaches were utilized. BLAST-P [24] services at NCBI and PDB database [25] were utilized; simultaneously Template Identification Tool from Swiss Institute of Bioinformatics was also utilized. Protein Homology/analogY Recognition Engine (PHYRE2) server [26] was employed for fold recognition.
Modeling studies
In a rational modeling strategy, it is recommended to include more than one template and methods to generate several models and then select best model from obtained results; rather than relying on single template and single method. Therefore both, standalone software like Modeller [27] as well as automated online SWISS MODEL server [28,29] were used in this study to generate the homology models.
Target-template alignment
The Align2D command [30] from Modeller was utilized to align query sequence to template structures. Align2D command is a modification of classic Needleman-Wunsch dynamic programming algorithm [31]. It considers structural information from the template when constructing an alignment, which is accomplished through a variable gap penalty function that tends to place gaps in solvent-exposed and curved regions, positions outside secondary structure segments, and between two positions that are close in space.
Model generation
Models for apo form of enzyme were generated by a special class derived from auto model command called as ‘Loop Model’. Loop-model class generates models by creating a new object for loop modeling which later builds standard comparative models utilizing the same approach of the auto model class and then refines loops in each of the created models. This methodology has an advantage that loops in models are refined scrupulously as compared to using auto model class. Five models were allowed to be built from each template and single loop model was generated after loop refinement process. Thus five models from each template were generated. Later on its cofactor (Mn ions), inhibitor and essential water molecules were inserted using SPDBV 3.7 [32]. This task was accomplished by separately loading the apo form of enzyme and cofactor (Mn ions), inhibitor, essential water molecules followed by merging these two separate layers into single molecule using commands available from “EDIT” menu of SPDBV 3.7.
Energy minimization
Energy minimization was carried out in vacuo using force field approach by means of GROMOS96 43Bl parameters set [33], without reaction field applying 20 steps of steepest descent algorithm within SPDBV 3.7.
Model evaluation
Best models were first short listed using QMEAN scoring function. Models were subject to evaluation using standard tools like ProCheck [12], WhatCheck/WhatIf [34] and Verify3D [35] Model was later visualized using Pymol. The resulting models of arginase were submitted to PMDB Database [36] and can be downloaded using PMDB ID, PM0078185.
Molecular dynamics of homology model
For further refinement and to verify thermodynamic stability of generated homology models, molecular dynamics simulations were carried out using CHARMM force field [37] implemented in NAMD [38] program. Initially the PDB-PSF pair of files was generated using ‘psfgen’ package implemented in VMD. The model was further solvated by placing it in water-box of suitable size utilizing ‘solvate’ package. Energy minimization was carried out for 2000 steps of conjugate gradient algorithm aiming to adjust the distribution of solvent molecule and to relax possible steric clashes that might be created, as coordinates of many atoms are guessed during generation of psf file. For all the simulations, periodic boundary conditions were applied in all cell dimensions. Long range interactions were dealt with using the switching function to consider electrostatic and van der Waal interactions by setting cutoff of 12.0 and switchdist of 8.0 angstrom. The SHAKE algorithm [39] was applied to constrain all bonds between hydrogen atoms and heavy atoms. Bonded and non-bonded forces were evaluated at every time step and full electrostatic forces were evaluated by application of the Particle Mesh Ewald (PME) method [40]. In the next stage, the temperature of the system was linearly increased from 0 K to 300 K within a time span of 300 ps. At each integration step, velocities were reassigned from a new Maxwell distribution and temperature was increased by 0.001 K. During the process of heating, the kinetic energy is pumped into the system among all degrees of freedom and hence it must be properly distributed. Thus, equilibration was carried out to equilibrate kinetic and potential energies. The system was equilibrated for over 1.5 ns at constant temperature of 300 K. During heating and equilibration stages, the integration time step was set to 1 fs. During equilibration, a frame was recorded to trajectory file after every 1 ps of simulation. Following this, in production stage of another 3 ns, all the dynamic behavior and structural changes of enzyme were analyzed by calculating energy of system and RMSD of trajectories.
Molecular docking
Docking analysis was carried out using two standard docking softwares that are available in public domain (ArgusLab and Autodock 4.2). Initially, 2D structures of the inhibitors were drawn in ChemDraw ® 8.0 (CambridgeSoft, Cambridge, MA, USA) and their SMILES were obtained. The 3D conformers of these compounds were then generated in SDF format using FROG2 Server [41].
Autodock 4.0 [42,43] implemented in Python Prescription 0.8 (PyRx) was used in docking analysis. The ligand molecules in sdf format were imported in PyRx environment via OpenBabel utility and were subject to energy minimization using UFF force field [39,44]; Conjugate gradient optimization algorithm was applied for over 200 steps while molecules were updated for every 1 step. Receptor molecule was prepared in traditional ‘pdbqt’ format after assigning charges to the receptor coordinate file by using ‘Make macromolecule’ command from Autodock menu. Auto-grid program was then utilized to obtain grid file with 69.3238, 56.7622, -3.8719 as x, y and z coordinates as grid centre. The affinity grid of 50 × 50 × 50 points was set using spacing of 0.375 angstrom in order to encompass entire active site. The Lamarckian Genetic Algorithm was used for the conformational search. Each Lamarckian job was set to have 10 runs. The initial population was restricted to 150 structures; while the maximum number of energy evaluation and generation were set to 27000. Single top individual was allowed to survive to next generation, rate of gene mutation was set to 0.02 and rate of crossover was set to 0.8; the rest of parameters were set as default values. The final structures were clustered according to native Autodock Scoring Function. The top ranked conformations of each ligand were selected (Table 1).
While using ArgusLab, the binding site was set in region where original inhibitor was placed in homology model. Dimensions of binding site bounding box were allowed to be automatically calculated. The same dimensions of box were used for docking remaining inhibitors so as to maintain consistency of chemical environment in active site. Grid resolution was set to 0.4 angstrom. The parameters required to calculate binding energies for the resulting docked structures were imported from Ascore.prm file since AScore function was used to score the individual binding poses. This parameter file holds all the coefficients for each term in the scoring function. Docking calculations were performed by selecting "ArgusDock" as the docking engine. An exhaustive search was performed by enabling “High precision” option in Docking precision menu, "Dock" was chosen as the calculation type. Ligand flexibility was allowed by selecting "flexible" option in the ligand menu. The docking poses showing lowest energy, maximum hydrogen bonding and minimum bumps with residues in active site were selected (Table 1).
Paired potential analysis
Further, paired potential analysis of docked conformers were carried out to scrutinize the per-atom score contribution of amino acid residues from model that are located around inhibitors in terms of ‘good’ and ‘bad’ potentials. These paired potential represent favorable and unfavorable interactions respectively. This task was achieved using an on-line program, DSX-ONLINE v0.88, knowledge-based DSX pair potentials that are based on the DrugScore potential [45]. The results were visualized in PyMol.
Results and Discussion
Previous attempts of arginse modeling have been made using computational approach to deduce the structure of arginase from H. pylori wild type strain 43504 [46]. Protein sequence alignment of arginase revealed the fact that sequence of arginase from H. pylori India 7 strain is different from wild type strain 43504. Difference of 5 percent in sequence identity was observed from that of wild type strain 43504 indicating considerable difference at primary structural level. Earlier model was generated using single template and single method; in contrast, we have used more than one template separately and employed three different methods to obtain more reliable models. Additionally, we have attempted to model entire sequence of protein in contrast of previous study that focused on modeling only central region of arginase.
Template identification and sequence analysis
From both the methods used for template search, three major structural hits of arginase were obtained from two species Thermus thermophilus having 26 % sequence identity and 90 % query coverage (PDB ID 2EF4, and 2EIV both with e-value of 7e-22), another was from Bacillus caldovelox also having 26 % sequence identity but covered only 87 % of query (PDB ID 1CEV with e-value of 2e-21). The comparison of e-values obtained from template identification results support usage of template from Bacillus caldovelox. Information about active site of arginase was acquired from SwissProt entry P53608 which correspond to arginase from Bacillus caldovelox. Two substrate binding regions are present in arginase of Bacillus caldovelox (124-HGDVN-128) and (135-SGN-137) along with few metal binding sites. In order to verify the fact that these regions are conserved in arginase from H. pylori, sequence of query/target and template were aligned. The substrate binding region 124-HGDVN-128 seems to be fairly conserved sequentially in H. pylori, same is the case with region 135-SGN-137 (Figure 1). Residues involved in metal binding is 178-D, which had replaced with another negatively charged amino acid E in H. pylori and can be considered as conserved as it creates same functional chemistry. 271-E is conserved. At position 240-T, although it seems that T is not conserved at this position but, it is to be noted that it forms a palindrome in another sequence, thus it might creates same chemical environment. All these facts suggest that structural model from Bacillus caldovelox can be used as template.
The accuracy of the structures generated by homology modeling is highly dependent on the sequence identity between target and template. Above 50% sequence identity, models tend to be reliable, with only minor errors in side chain packing. Below 30% identity, serious errors occur, sometimes resulting in the basic fold being mis-predicted. When sequence identity between template and query sequence falls in range of 20 to 30 %, it is said that sequence alignment is in "Twilight Zone" where one is uncertain if two sequences are indeed homologous. In such situations fold recognition approach is utilized to identify a suitable fold that can be used as template. Since arginase sequence from H. pylori shares only 26 % sequence identity with its homolog from Bacillus caldovelox and falls in “Twilight Zone”, an alternative method of fold recognition was additionally employed in order to identify suitable fold. Top result from fold recognition comprises of a model based on fold named “D1PQ3A” belonging to ‘Arginase-like amidino hydrolases’ family and ‘Arginase/deacetylase’ super-family and fold respectively; claiming 100 % confidence with 92% query coverage. Furthermore, fold D1PQ3A correspond to PDB entry 1PQ3 which describes structure of arginase from Human. Thus two templates (one from Bacillus caldovelox and another obtained from fold recognition result were used in this study for building a reliable structural model.
Target template alignment
The pair-wise alignment of query sequence to two templates showed local differences with respect to gap positions. Four gaps are clearly observed in alignment of query with template from Bacillus caldovelox; two gaps of length 2 and remaining two of length 16 and 11 residues that form a loop at position 107-DK-108, 154-SEEKAWQKLCSLGLEK-169, 289-KH-290 and terminal 312-KDKKPSFAMSY-322. When fold obtained from fold recognition method was used as template, gaps at position 107-108 and 289-290 were completely removed and the remaining two loops were reduced to 13 and 6 residues respectively. These changes significantly improved the model obtained using human arginase as template. Chances of getting better models are increased if there are fewer gaps in alignment.
Modeling
Topology diagram for model was obtained using Pro-Origami server [47]. The models obtained were composed of 10 helices (H1-H10), 9 loops (L1-L9) and characteristic beta sheet composed of 8 strands. Structural models obtained from Modeller showed typical 3 layers of secondary structure arrangement formed as α-helix/β-sheet/ α -helix which is signature of ‘Arginase/deacetylase’ fold. The parallel beta-sheet is composed of 8 strands having order 2-1-3-8-7-4-5-6, which again signifies that protein has adopted the desired fold.
Model Evaluation
QMEAN
All the 12 models were ranked on QMEAN Server [48] using QMEAN Scoring Function [48] on basis of both global and local error estimates taking into account of C-beta interaction energy, all atom pair-wise energy, solvation energy, torsion angle energy, secondary structure agreement and solvent accessibility agreement. The sole purpose to use QMEAN score in this study was to pick out good models from the set of produced results. Three out of five structures created using fold derived from human arginase illustrated score above 6, while none of the model produced using template from Bacillus caldovelox even approached this score indicating that models produced using hybrid approach of fold-recognition and then using Modeller performs better than just relying on Modeller to produce homology models. It is also evident that models produced by automated online server SWISS MODEL showed poor QMEAN score despite the template used to generate them. From this observation it can be concluded that, in present study, better results are obtained using stand alone approach of generating models using Modeller, as compared to automated online approach.
ProCheck
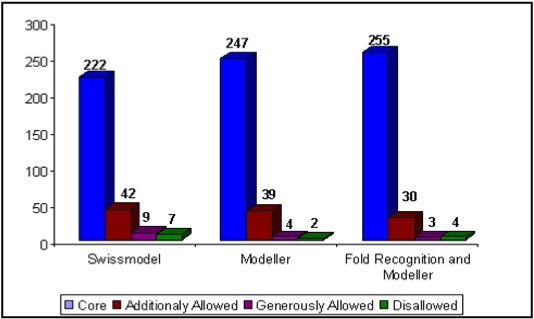
Comparative evaluation study of model created using ‘fold recognition and Modeller’ approach along with best model produced using template from Bacillus caldovelox and SWISS-Model was carried out on basis of geometrical and stereo-chemical constraints from ProCheck. Table 2 along with Figure 2 illustrate that best model created using template from fold recognition employing Modeller program has more than 87 percent of its non-proline and non-glycine amino acid residues occupied in core region, 10.3 percent in additionally allowed region, one percent in generously allowed region and 1.4 % in disallowed region as compared to model created using template from Bacillus caldovelox employing Modeller has 84.6 % in core region, 13.4 in additionally allowed region, 1.4 % in generously allowed region, 0.7 % in disallowed region; while best model build using SWISS MODEL poorly performed having only 79.3 % of its non-proline and non-glycine amino acid residues in core region, 15 % in additionally allowed region, 3.2 in generously allowed region and 2.5 % in disallowed region. In other words more than 20 % of non-proline and non-glycine residues fall out of core region when SWISS MODEL was used to generate model for arginase as compared to 15 % of same type of residues lie outside core region when template from Bacillus caldovelox was used to generate models using Modeller; while using hybrid approach of fold recognition and Modeller, it was found that only 12 % of non-proline and non-glycine residues fall out of core region. These figures again demonstrate that model generated using hybrid approach stand better in terms of geometrical and stereo chemical properties. Residues 40-Q, 89-S, 106-K and 156-D are found to be present in disallowed region; these residues are not really a matter of concern as their positions are happened to be present in region that is remote from active site [49].
Verify3D
The compatibility of an atomic model (3D) with its own amino acid sequence (1D) was analyzed using Verify3D program. Residues scoring value above 0.2 are considered to have better compatibility with its structure. More than 76 % of residues in proposed model stay well above threshold score, once more demonstrating good quality of model. However, there is patch of sequence (38-G to 65-F) that exhibited negative score, this fragment is completely exposed to solvent and do not play any role in stability. Moreover, amino acid residues constituting active site have acquired score in range of 0.4 to 0.6; these numbers point out those residues in active site are absolutely compatible with their surroundings.
WhatIf / WhatCheck
Features affecting quality of models like tau angle problems, side chain planarity problems, connections to aromatic rings out of plane and side chain flips etc were analyzed using WhatCheck/WhatIf suites. Best models obtained using Modeller and SWISS MODEL was subject to test using 82 parameters provided in WhatIf version 20111205-2247. Structures created using Modeller passed most of the tests, except one ‘side chain flips’ and one ‘tau angle problem’ were detected. These abnormalities along with few ‘abnormally short inter atomic distances’ were found to be present in the template structure and consequently were inherited in proposed model. On the other hand, four severe problems in model created using SWISS MODEL were observed. In brief, best model generated by Modeller using template of fold derived from human arginase possess the finest geometrical and stereo-chemical properties, its coordinates are highly compatible with sequence and violates minimally in WhatIf analysis, therefore it was further utilized in molecular dynamics and docking investigation.
Active site of Arginase
Active site of arginase is found to be highly conserved and the role of metal ions is extremely important in catalysis as it is known that metal ions lower the pKa for proton ionization and activate a coordinated water molecule, and further create the hydroxide ion that nucleophilically attacks the guanidino carbon of the scissile bond of l-arginine [50-52]. In present model of arginase of H. pylori, the binuclear Mn(II) center is found to be situated at the bottom of an 17 Å-deep active site cleft which is close to experimentally verified depth of 15 Å. Sheets 4, 5 and 7 seem to form base of active site and is surrounded by loops and helices forming supportive walls.
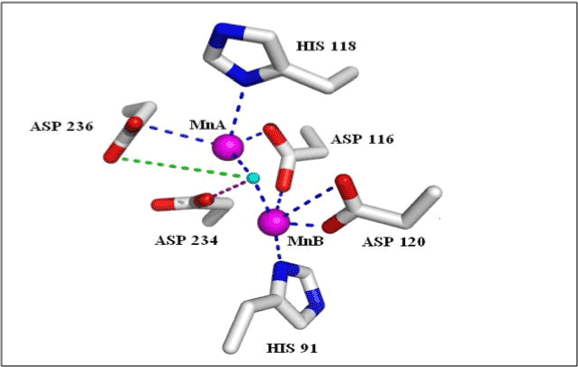
According to a detailed comparative study of more than 30 experimentally solved structures of arginase family, it has been found that numerous aspartate and histidine residues in Mn binding region are highly conserved. These residues are considered to play an imperative role in binding and catalysis of substrate [53]. MnA is known to possess square pyramidal coordination (Figure 3 blue lines); MnA (magenta sphere) is coordinated with nitrogen of 118-H and one of oxygen 236-D. These two residues thus play role of major terminal ligands while one oxygen of bidentate 116-D and additional oxygen from monodentate 234-D along with hydroxide ions (cyan sphere) act as its bridging ligands.
On the other hand, MnB (magenta sphere) is identified to be coordinated with distorted octahedral geometry (blue lines) by nitrogen and oxygen of terminal ligands 91-H and bidentate 120-D respectively. Oxygen of bidentate 116-D, monodentate 234-D along with hydroxide ion (cyan sphere) forms its bridging ligands. A hydrogen bond is donated by metal-bridging hydroxide to the non-coordinating oxygen of 236-D. All these interactions are found to be essentially present in experimentally verified active site of arginases [54].
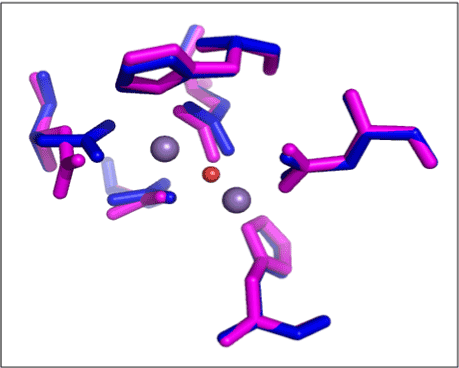
Accuracy of the obtained model was checked by performing its structural alignment with structure obtained by x-ray crystallography. The homology model and model of human arginase (1PQ3) were superimposed and RMSD of residues involved in active site was calculated (Figure 4). Since the fold that was used as a template to derive structural model for H. pylori was originated from human arginase, hence it was used as standard for comparison. The RMSD value for all atom superimposition of active site residues was found to be as low as 0.55 Å indicating very high accuracy of H. pylori active site model.
Loop study
H. pylori arginase contains two 13 and 6-residue loops inserted within loop L5 and at c-terminal end. As these loops are located far away from active site and it is already known that c-terminal polypeptide mediates more than 50% of inter monomer contact surface area [50], the functional role of these loops can be hypothesized to be involved in stabilization of oligomeric structure.
Molecular dynamics
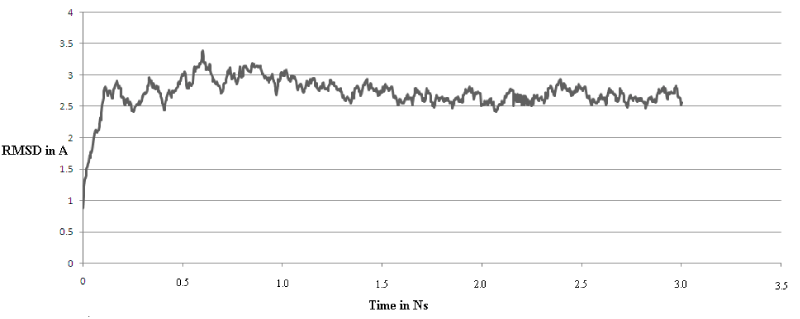
Molecular dynamics simulation was carried out in order to verify that the model obtained from homology modeling is thermodynamically and conformationally stable at physiological conditions (in solvent at 300 K). RMSD analysis of the trajectories with respect to the starting structure revealed sharp increase in RMSD from 0 to approximately 2.3 Å during first 800 ps; after 1.5 ps RMSD value was restored around 2.5 Å that did not changed significantly till completion of simulation as it is shown in (Figure 5). Hence, thermodynamically and conformationally stable molecule of arginase was obtained around 300 K at near physiological conditions. Comparison of model before and after molecular dynamics simulations revealed slight movement in peripheral domains, but negligible differences were recorded in active site. This observation along with RMSD values indicate that applied time for simulation was enough to obtain an equilibrium structure of arginase from H. pylori.
Docking and paired-potential analysis
Autodock uses a stochastic grid-based methodology that employs the genetic algorithm to sample different populations of ligand conformations in their binding to the receptor. In this method each and every calculated pose is energetically evaluated by a series of energy minimization steps, in which unsuccessful docking results are discarded. The genetic algorithm is a widely used and reliable algorithm but it has some limitations (Park et al., 2006), the most significant includes the possibility of the optimization of the ligand conformations getting trapped in local minima, this fact might explain poor prediction of binding energy in case of some ligands in our study, that are known to be good inhibitors experimentally (in case of ligand L-NNA in Table 1). ArgusLab on the other hand, provides both algorithms, first, the stochastic search which is similar to the genetic algorithm provided by Autodock, and second includes an exhaustive search method based on identification of complementary shapes of the ligand and the receptor, referred to as "ShapeDock" or "ArgusDock". Hence results obtained using both the software simultaneously will be more reliable as compared to individual usage of either of the software.
The validity of a docking system can be checked by docking the experimentally verified pose back into the receptor. This procedure follows the rationale that a good docking engine should replicate the experimental binding modes of the ligand. After docking, root mean square deviation (RMSD) value of the predicted pose to experimentally verified pose is calculated. RMSD value indicates the measure of spatial similarity between two structures. If the RMSD value is found to be less than certain value (typically <2.00 angstrom), the prediction of binding mode is considered as successful. RMSD value of 0.62 angstrom was observed while docking experimentally verified ligand (lysine) into its own active site using AutoDock. This value indicates that predicted binding mode is nearly identical to the X-ray crystallography conformer.
According to a study, two histidines and four aspartate residues are essentially required for providing stability and catalysis of the binuclear metal center [55]. In the proposed model 91-H, 118-H, 116-D, 120-D, 234-D and 236-D are found to be structurally conserved and play an important role in inhibition. Besides these residues, here we report, three more novel interactions never reported earlier. Our data suggest the possibility of these residues to play an important role in modulation of arginase activity. According to paired potential calculation on DSX server, these residues have shown to interact favorably with inhibitors. Favorably interacting atoms from protein are shown to be surrounded by green spheres, while the sizes of the spheres correspond to the values of the contributing per-atom scores. Docking of ornithine to active site of arginase demonstrates the importance of water molecules is stabilization by forming a bridge network of hydrogen bond with terminal NH2 group of ornithine and NE2 atom of 133-H. While another network participating in stabilization of active site includes NE2 atom of 122-H donating hydrogen bond to carboxyl oxygen that bridges via a water molecule to NH2 group attached to c-alpha carbon atom. The same NH2 group is also involved in formation of hydrogen bond with OD1of 120-D. Favorable DSX potential contribution (green spheres) of the same 122-D, 133-H and 120-D residues is depicted in adjacent figure.
The crucial role of hydroxyl-guanidine fragment of N omega hydroxyl L arginine inhibitor is evident from its docking to arginase active site. The terminal oxygen of hydroxyl-guanidine moiety stabilizes the active site by forming hydrogen bond with NE2 atom of 133-H. Another nitrogen of the same moiety takes part in series of hydrogen bond bridge that involve its coordination with water molecule and OD1 atom of 120-D. The same water molecule continues to bridge with oxygen of carboxyl group of inhibitor those in-turn hydrogen bonds with NE2 atom of His-122. ND1, NE2 atoms of 122-H along with NE2 atom of 133-H and OD1, CG and CB atoms of 120-D seems to have major favorable DSX potential contribution towards three terminal carbons on inhibitor (magenta spheres).
The nitrogen of guanidino group of N-alpha-L-acetyl-arginine inhibitor seems to form a hydrogen bond with water. Additional water is involved in series of hydrogen bonding network that includes oxygen of carboxyl group from inhibitor hydrogen bonding with NE2 of 133-H via nitrogen of NH group on inhibitor. NE2 atom of 118-H hydrogen bonds with carboxyl oxygen of inhibitor, that seems to confer stability to enzyme–inhibitor complex. Same atoms are also observed to have favorable DSX potential contribution towards inhibitor as obtained from DSX server. Moreover, our docking study coincides with experimental values in case of SNAP which showed poor binding affinity towards arginase (-2.95 Kcal/mol in Autodock) as compared to other arginase inhibitor [56].
Series of chemically diverse small molecule inhibitors like fluoride (IC50, 1500 µM), L-valine (IC50, 650 µM), Nω-hydroxy-L-arginine (IC50, 20 µM), Nω-hydroxy-nor-L-arginine (IC50, 0.8 µM), 2(S)-amino-6-boronohexanoic acid (ABH, IC50 1.6 µM), dehydro ABH (IC50, 15 µM) etc. have been tested in wet-lab experimental settings and demonstrated significant inhibition of arginase activity [56]. As there is clear evidence of arginase inhibition using small molecule inhibitors, the results of the present study may help in design and development of novel arginase inhibitors of H. pylori.
Conclusion
The basic objective of the present investigation was to generate thermodynamically as well as conformationally stable model of arginase from H. pylori which would be reliable, both chemically as well as biologically; we have successfully developed it. We would also conclude that in general, models generated from hybrid approach implementing methods like fold recognition and satisfaction of spatial restrains gives more accurate results as compared to individual usage of either of the methods. The model so produced is valid not only in terms of active site which is highly similar to crystal structure of enzyme, but also adopts the overall topology of the ‘arginase/deacetylase’ fold. The proposed structural model of arginase has successfully passed geometrical and stereo-chemical tests; its atomic coordinates are fully compatible to its sequence and have minimally violated parameters from ‘WhatCheck’ suite indicating its best quality. Docking studies using two software and paired potential analysis have revealed newer interactions involving residues that have never been reported in earlier studies and thus has enormous scope to be utilized and verified experimentally for finding novel class of candidate drugs against H. pylori.
Acknowledgements
The authors are thankful to Department of Science and Technology (DST), New Delhi, India for financial assistance under Fast Track Scheme for Young Scientist (ST/FT/CS-012/2009).
References
- Labigne A, de Reuse H (1996) Determinants of Helicobacter pylori pathogenicity. Infect Agents Dis 5: 191-202. [Crossref]
- McColl Ke (1997) Helicobacter pylori: clinical aspects. J Infect 34: 7-13. [Crossref]
- Laurila A, Bloigu A, Näyhä S, Hassi J, Leinonen M, et al. (1999) Association of Helicobacter pylori infection with elevated serum lipids. Atherosclerosis 142: 207-210. [Crossref]
- Helicobacter and Cancer Collaborative Group (2001) Gastric cancer and Helicobacter pylori: a combined analysis of 12 case control studies nested within prospective cohorts. Gut 49: 347-353. [Crossref]
- Malfertheiner P, Megraud F, O'Morain C, Bazzoli F, El-Omar E, et al. (2007) Current concepts in the management of Helicobacter pylori infection: the Maastricht III Consensus Report. Gut 56: 772-781. [Crossref]
- Chey WD, Wong BC; Practice Parameters Committee of the American College of Gastroenterology (2007) American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol 102: 1808-1825. [Crossref]
- Gürbüz AK, Ozel AM, Yazgan Y, Günay A, Polat T (2003) Does eradication of Helicobacter pylori reduce hypergastrinaemia during long term therapy with proton pump inhibitors? East Afr Med J 80: 150-153. [Crossref]
- DiBaise JK (1999) PPIs, H. pylori, and atrophic gastritis: putting the issue to rest? Am J Gastroenterol 94: 3403. [Crossref]
- Zullo A, Hassan C, Morini S (2002) Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 346: 65-67. [Crossref]
- Kuipers EJ (2006) Proton pump inhibitors and gastric neoplasia. Gut 55: 1217-1221. [Crossref]
- Waldum HL, Qvigstad G (2007) Proton pump inhibitors and gastric neoplasia. Gut 56: 1019-1020. [Crossref]
- Houben MH, van de Beek D, Hensen EF, de Craen AJ, Rauws EA, et al. (1998) A systematic review of Helicobacter pylori eradication therapy--the impact of antimicrobial resistance on eradication rates. Aliment Pharmacol Ther 13: 1047-1055. [Crossref]
- Gisbert JP, Gonzalez L, Calvet X (2005) Systematic review and meta-analysis: proton pump inhibitor vs. ranitidine bismuth citrate plus two antibiotics in Helicobacter pylori eradication. Helicobacter 10: 157-171. [Crossref]
- Mendz GL, Hazell SL (1996) The urea cycle of Helicobacter pylori. Microbiology 142: 2959-2967. [Crossref]
- Mendz GL, Holmes EM, Ferrero RL (1998) In situ characterization of Helicobacter pylori arginase. Biochim Biophys Acta 1388: 465-477. [Crossref]
- Marshall BJ, Barrett LJ, Prakash C, McCallum RW, Guerrant RL (1990) Urea protects Helicobacter (Campylobacter) pylori from the bactericidal effect of acid. Gastroenterology 99: 697-702. [Crossref]
- Sjöström JE, Larsson H (1996) Factors affecting growth and antibiotic susceptibility of Helicobacter pylori: effect of pH and urea on the survival of a wild-type strain and a urease-deficient mutant. J Med Microbiol 44: 425-433. [Crossref]
- Johnson MS, Srinivasan N, Sowdhamini R, Blundell TL (1994) Knowledge-based protein modeling. Crit Rev Biochem Mol Biol 29: 1-68. [Crossref]
- Meshram RJ, Gavhane A, Gaikar R, Bansode Ts, Maskar A, et al. (2010) Sequence analysis and homology modeling of laccase from Pycnoporus cinnabarinus. Bioinformation 5: 150-154. [Crossref]
- Huynh NN1, Harris EE, Chin-Dusting JF, Andrews KL (2009) The vascular effects of different arginase inhibitors in rat isolated aorta and mesenteric arteries. Br J Pharmacol 156: 84-93. [Crossref]
- Selamnia M, Mayeur C, Robert V, Blachier F (1998) Alpha-difluoromethylornithine (DFMO) as a potent arginase activity inhibitor in human colon carcinoma cells. Biochem Pharmacol 55: 1241-1245. [Crossref]
- Neudert G, Klebe G (2011) DSX: a knowledge-based scoring function for the assessment of protein-ligand complexes. J Chem Inf Model 51: 2731-2745. [Crossref]
- Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, et al. (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31: 365-370. [Crossref]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. (1990). Basic local alignment search tool. J Mol Biol 215: 403-410. [Crossref]
- Westbrook J, Feng Z, Chen L, Yang H, Berman HM (2003) The Protein Data Bank and structural genomics. Nucleic Acids Res 31: 489-491. [Crossref]
- Kelley LA, Sternberg MJ (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4: 363-371. [Crossref]
- Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779-815. [Crossref]
- Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res 31: 3381-3385. [Crossref]
- Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22: 195-201. [Crossref]
- Madhusudhan MS, Marti-Renom MA, Sanchez R, Sali A (2006) Variable gap penalty for protein sequence-structure alignment. Protein Eng Des Sel 19: 129-133. [Crossref]
- Needleman SB, Wunsch CD (1970) A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48: 443-453. [Crossref]
- Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling. Electrophoresis 18: 2714-2723. [Crossref]
- Van Gunsteren WF, Brunne RM, Gros P, van Schaik RC, Schiffer CA, et al. (1994) Accounting for molecular mobility in structure determination based on nuclear magnetic resonance spectroscopic and X-ray diffraction data. Methods Enzymol 239: 619-654. [Crossref]
- Hooft RW, Vriend G, Sander C, Abola EE (1996) Errors in protein structures. Nature 381: 272. [Crossref]
- Eisenberg D, Lüthy R, Bowie JU (1997) VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol 277: 396-404. [Crossref]
- Castrignanò T, De Meo PD, Cozzetto D, Talamo IG, Tramontano A (2006) The PMDB Protein Model Database. Nucleic Acids Res 34: D306-309. [Crossref]
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102: 3586-3616. [Crossref]
- Phillips JC1, Braun R, Wang W, Gumbart J, Tajkhorshid E, et al. (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26: 1781-1802. [Crossref]
- Rappe AK, Colwell KS, Casewit CJ (1992) Application of a Universal Force Field to Organic Molecules. J Am Chem Soc 114: 10035-10046.
- Darden T, York D, Pedersen L (1993) Particle mesh Ewald: An N . log(N) method for Ewald sums in large systems. J Chem Phys 98:10089-10092.
- Miteva MA, Guyon F, Tufféry P (2010) Frog2: Efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Res 38: W622-627. [Crossref]
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, et al. (1998) Automated Docking Using a Lamarckian Genetic Algorithm and and Empirical Binding Free Energy Function. J Comput Chem 19: 1639-1662.
- Huey R, Morris GM, Olson AJ, Goodsell DS (2007) A semiempirical free energy force field with charge-based desolvation. J Comput Chem 28: 1145-1152. [Crossref]
- Gohlke H, Hendlich M, Klebe G (2000) Knowledge-based scoring function to predict protein-ligand interactions. J Mol Biol 295: 337-356. [Crossref]
- Benkert P, Tosatto SC, Schomburg D (2008) QMEAN: A comprehensive scoring function for model quality assessment. Proteins 71: 261-277. [Crossref]
- Azizian H, Bahrami H, Pasalar P, Amanlou M (2010) Molecular modeling of Helicobacter pylori arginase and the inhibitor coordination interactions. J Mol Graph Model 28: 626-635. [Crossref]
- Stivala A, Wybrow M, Wirth A, Whisstock JC, Stuckey PJ (2011) Automatic generation of protein structure cartoons with Pro-origami. Bioinformatics 27: 3315-3316. [Crossref]
- Wlodawer A, Minor W, Dauter Z, Jaskolski M (2008) Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. FEBS J 275: 1-21. [Crossref]
- Kanyo ZF, Scolnick LR, Ash DE, Christianson DW (1996) Structure of a unique binuclear manganese cluster in arginase. Nature 383: 554-557. [Crossref]
2021 Copyright OAT. All rights reserv
- Christianson DW1, Cox JD (1999) Catalysis by metal-activated hydroxide in zinc and manganese metalloenzymes. Annu Rev Biochem 68: 33-57. [Crossref]
- Ash DE, Cox JD, Christianson DW (2000) Arginase: a binuclear manganese metalloenzyme. Met Ions Biol Syst 37: 407-428. [Crossref]
- Perozich J, Hempel J, Morris SM Jr (1998) Roles of conserved residues in the arginase family. Biochim Biophys Acta 1382: 23-37. [Crossref]
- Christianson DW, Alexander RS (1989) Carboxylate-histidine-zinc interactions in protein structure and function J Am Chem Soc 111:6412-6419.
- Ouzounis CA, Kyrpides NC (1994) On the evolution of arginases and related enzymes. J Mol Evol 39: 101-104. [Crossref]
- Wei LH, Wu G, Morris SM Jr, Ignarro LJ (2001) Elevated arginase I expression in rat aortic smooth muscle cells increases cell proliferation. Proc Natl Acad Sci U S A 98: 9260-9264. [Crossref]
- Cama E, Pethe S, Boucher JL, Han S, Emig FA, et al. (2004) Inhibitor coordination interactions in the binuclear manganese cluster of arginase. Biochemistry 43: 8987-8999. [Crossref]