Abstract
Memantin, a non-competitive NMDA receptor antagonist, is administered to patients with Alzheimer’s disease (AD) to expect partial blocking of NMDA channels without dramatically affecting normal transmission of this channel. Recent studies have shown results that memantine can affect the amyloid precursor protein (APP), amyloid beta protein (Aβ), and tau metabolism. However, the molecular mechanism underlying memantine-induced neuroprotective effect remain unclear. In this study, we investigated the neuroprotective effects of memantine in neurons in vitro. Primary neuronal cultures incubated with Aβ1-42 (3 μM) for 48 h exhibited neuronal death, as assayed by LDH release and Calcein AM/PI staining. The Aβ1-42-induced neuronal death was prevented by the addition of memantine at concentrations of 1-10 μM. MK801, a competitive inhibitor of NMDA channel, also inhibited neuronal death caused by Aβ1-42 (3μM). There was no additive effect of memantine and MK801 when they were administered together. These results suggest that memantine attenuates neurotoxicity caused by Aβ1-42 via its NMDA channel blockade, and that beneficial effects of memantine on AD symptoms may be partially explained by its neuroprotective effects against Aβ.
Key words
Memantine, Alzheimer’s disease, Aβ toxicity, NMDA channel
Introduction
One of the neuropathological hallmarks of Alzheimer’s disease (AD) is the formation of extracellular amyloid deposits [1]. The major component of these amyloid deposits is the 39- to 42-aa peptide of the amyloid Aβ-protein (Aβ) [2,3]. One of the species of Aβ, which terminates at residue 40 with a C-terminus (Aβ1-40), is the predominant soluble species in biological fluids, such as cerebrospinal fluid. The longer form of Aß, which ends at residue 42 (Aβ1-42), accumulates and is predominantly found in parenchymal plaques [4,5]. Aβ1-42 is normally produced and secreted by cells in much lower quantities than Aβ1-40, which comprises approximately 90% of the total amount of secreted Aβ. It is thought that aggregated Aβ is neurotoxic and can initiate the progression of AD pathophysiology [6,7]. Aβ oligomers also play an important role in AD pathogenesis [8,9]such that neurodegeneration can be induced in the mouse brain without amyloid plaque formation [9].We previously reported that Aβ1-42 is highly amyloidogenic and causes neurotoxicity, while Aβ1-40 remains in a monomer form, protecting neurons from metal-dependent free radical generation [10].
Memantine is a non-competitive NMDA receptor antagonist, with moderate binding affinity and rapid receptor kinetics [11,12]. These pharmacological properties enable memantine to be administered to patients with AD, partially blocking NMDA channels without dramatically affecting its neurotransmission. In patients with AD, memantine can delay the progression of memory impairments and attenuate AD-related symptoms, including visual hallucinations, agitation, and delirium. However, it is not completely understood whether memantine’s beneficial effects on AD symptomology can be solely explained by this mechanism. Recent studies have shown that in addition to preventing AD symptoms and improving AD-related memory impairments in AD mouse models, memantine attenuates the Aβ burden in neuronal cultures and in the APP transgenic (Tg) mouse [13-16]. Previous reports have demonstrated that memantine treatment at clinically relevant concentrations reduced levels of soluble Aβ oligomers, fibrillar Aβ oligomers, and Aβ depositions in APP Tg mice [13,15,17]. In addition, memantine treatment also improved cognition and reduced phosphorylated tau levels in APP Tg mice [13,15]. Memantine rescued soluble Aβ-induced short-term memory deficits in a rat model[18] and improved spatial learning and memory impairments in APP/PS1 Tg mice via NGF signaling[19].
In parallel with these in vivo data, memantine treatment reduced the generation of Aβ42 and the Aβ42/Aβ40 ratio in cultured neurons [16]. These data suggest that memantine may be a disease-modifying drug for AD by altering Aβ metabolism in the brain. However, the molecular mechanisms underlying memantine-induced neuroprotective effects are not fully understood. In this study, we investigated the neuroprotective effects of memantine on Aβ42-induced neurotoxicity in neurons.
Materials and methods
Materials
Memantine was obtained from Dai-ichi Sankyo Co. Ltd. (Tokyo, Japan). Memantine was dissolved in distilled water (DW) at a concentration of 100 mM to make aliquots, which were stored at -80 C until use. Memantine in each aliquot was diluted in the culture medium to a concentration of 500 μM and was added into the neuron cultures to the final concentrations used for the experiments. MK801 was purchased from Sigma Aldrich Japan (Tokyo, Japan), and was dissolved in DW at a concentration of 5 mM. The aliquots were made and stored at -80°C until use. Each aliquot was further diluted to 200 μM in the culture medium and was added into the neuron cultures to make a final concentration used for the experiments.
Cell culture
All experiments were performed in compliance with existing laws and institutional guidelines. Neuron-rich cultures were prepared from cerebral cortices as previously described [10], with some modifications. In brief, uteri of gravid rats at embryonic days 17-18 were removed under anesthesia. Cerebral cortices from fetal rat brains were dissected, freed of meninges and diced into small pieces; the cortical fragments were incubated in 0.25% trypsin and 20 mg/ml DNase I in phosphate-buffered saline (PBS) (8.1 mM Na2HPO4, 1.5 mM KH2PO4, 137 mM NaCl and 2.7 mM KCl, pH 7.4) at 37°C for 20 min. The fragments were then dissociated into single cells by pipetting. The cells were suspended in the feeding medium and plated onto poly-D-lysine-coated 12-well plates at a cell density of 2 × 105/cm2. The feeding medium consisted of Dulbecco's modified Eagle’s medium nutrient mixture (DMEM/F12; 50%: 50%) and N2 supplements. Over 99% of the cultured cells were identified as neurons by immunocytochemical analysis using monoclonal antibody against microtubule associated protein 2, a neuron-specific marker, at 3 days in culture.
Preparation of Aβ
Synthetic Ab1-42 (TFA salt) was purchased from Peptide Institute Inc. (Osaka, Japan). Aβ1-42 was dissolved in dimethyl sulfoxide (DMSO) at 13.3 mM, and diluted with PBS to obtain a 350 µM stock solution.
Viability assay
The release of the cytoplasmic enzyme, lactate dehydrogenase (LDH), into culture medium was determined for the quantification of cell death. Fifty microliters of culture medium were transferred to a fresh 96-well flat bottomed plate and a colorimetric LDH-release assay was performed according to the instructions of the manufacturer (Promega, WI), and absorbance were read at 490 nm immediately thereafter. For determination of total LDH, the neuronal cultures were incubated with 100 mM H2O2 for 10 min at room temperature and released LDH was determined, and the percentage of released LDH per total LDH in each culture was calculated. Calcein-AM and Propidium Iodide (PI) staining kit (Dojindo Molecular Tech, Inc., Maryland, USA) was used for the determination of live and dead cells in culture.The staining was performed according to the cell staining protocol in the manual by the company.
Microphotographs
The cultured neurons were taken photographs using Keyence B2-8000 with objective lens x 20. The living and dead cells in each field were counted, and the ratio of live/total cells was determined.
Statistical analysis
Statistical analysis was performed using StatView computer software (Macintosh), and multiple pairwise comparisons among the sets of data were performed using analysis of variance (ANOVA) and the Bonferroni t-test.
Results
The primary neuron cultures were prepared form rat embryo brain as described under the Materials and Methods. To determine the effect of memantine on Aβ-induced neurotoxicity, cultured neurons were exposed to Aβ1-42 at a concentration of 3 μM in the presence of memantine at 0, 1, 2, or 5 μM. Forty-eight h after the commencement of incubation, LDH released into the conditioned media was determined. The time-dependent effect of memantine on Aβ1-42-inducing neurotoxicity was shown in Figure1. Aβ1-42 treatment,at a concentration of 3 μM, caused neuronal cell death in a time-dependent manner, and memantine treatment attenuated this cell death(Figure1).
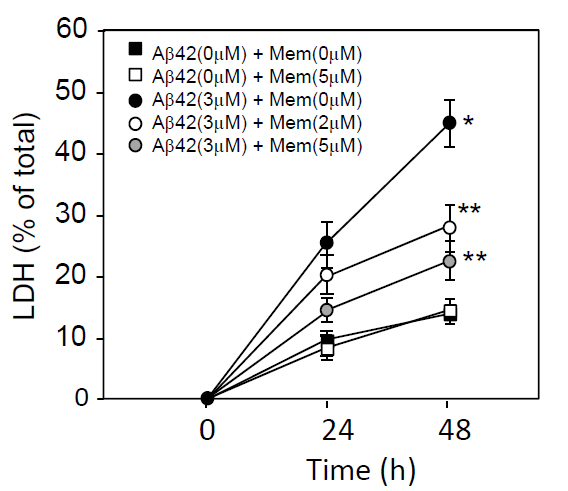
Figure 1. Time-dependent LDH release from cultured neurons treated with Aβ1-42 and/or memantine. Neuronal cultures were prepared from embryonic rat brains. Neurons were cultured in DMEM containing an N2 supplement for 2 days, washed in DMEM 3 times, and incubated in DMEM containing an N2 supplement in the presence or absence of 3 μM Aβ1-42 plus memantine (0, 2, or 5 μM). Twenty-four and 48 h later, the culture media was harvested and used for the LDH release assay, as described in the Materials and Methods section. Data are presented as the mean ± SD. N=3 per group. Three independent experiments had similar results. *P<0.01 and **<0.05 vs. control.
Figure 2 shows the ratio of LDH released into the conditioned media per total LDH in the neuron culture treated with varying concentrations of memantine in the presence or absence of Aβ1-42 (3 μM). Cell death, as shown as LDH release, was induced by Aβ1-42 treatment and this cell death was inhibited by the addition of memantine in a dose-dependent manner. Interestingly, cell death induced under the culture conditions was also prevented by the addition of memantine, suggesting that memantine prevented cell damage induced not only by Aβ, but also by culture itself.
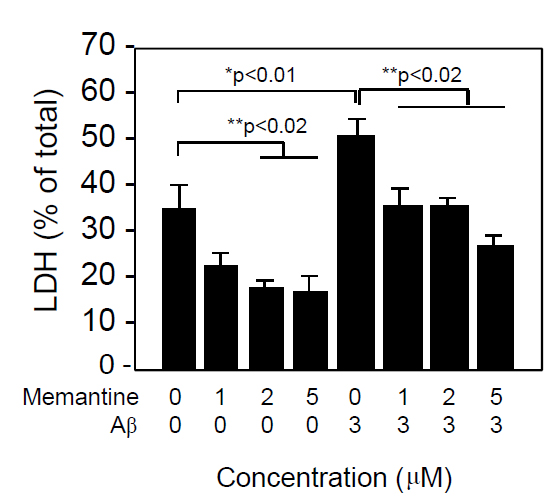
Figure 2. Memantine prevented Aβ1-42-induced neuronal death in a dose-dependent manner. Neuronal cultures were prepared from embryonic rat brains as described in the Materials and Methods section. Neurons were cultured in DMEM containing an N2 supplement for 2 days, washed in DMEM 3 times, and incubated in DMEM containing an N2 supplement in the presence of Aβ1-42 and memantine at the indicated concentrations. Forty-eight hours later, the culture media was harvested and used for the LDH release assay, as described in the Materials and Methods section. Data are presented as the mean ± SD. N=3 per group. Three independent experiments had similar results. *P<0.01 and **<0.02 vs. control.
We further determined cell survival and/or cell death by another assay system using Calcein AM and PI staining (Figure 3). Similarly to the results assayed by LDH release, 3 μM Aβ1-42 induced neuronal cell death, and 5μM memantine alone did not caused cell death. Aβ1-42-induce neural cell death was attenuated in the presence of memantine at 5μM. Based on the Calcein AM and PI staining, the survival ratio and the dead ratio per total cells were determined by counting Calcein AM-positively stained cells and PI-positively stained cells. Memantine did not have any effect on cell survival, when it was treated alone (Figure 4A), and it significantly prevented neuronal cell death induced by Ab1-42 (Figure 4B).
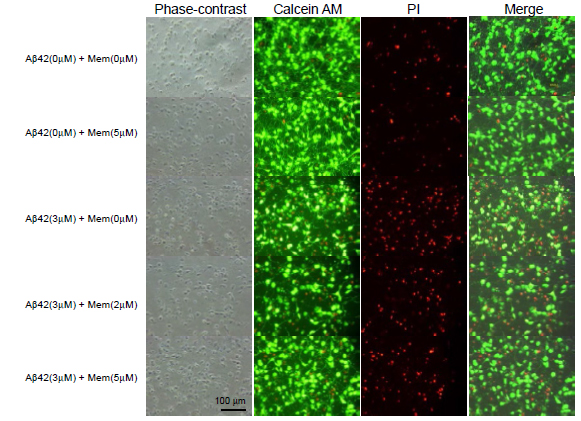
Figure 3. Cell survival was determined using Calcein AM and PI staining. Cultured neurons were treated with memantine (0, 2, or 5 μM) in the presence (B) or absence (A) of Aβ1-42 (3 μM) for 48 h. The cells were stained with Calcein AM and PI and imaged using a CCD camera.
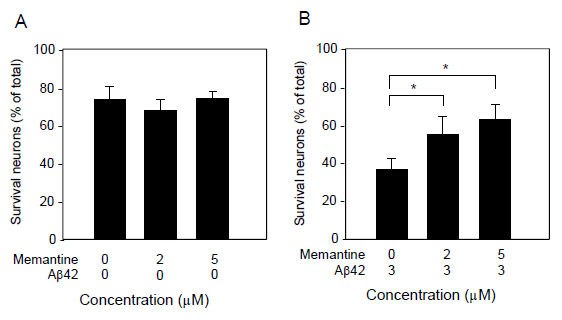
Figure 4. Quantification of neuronal survival as determined by Calcein AM and PI staining. Cultured neurons were treated with memantine (0, 2, and 5 μM) in the presence (B) or absence (A) of Aβ1-42 (3 μM) for 48 h, and then stained with Calcein AM and PI. The cultures were imaged with a CCD camera. The number of Calcein AM-positive and PI-positive cells were counted in randomly selected fields. Data are presented as the mean ± SD. N=3 per group. *P<0.02.
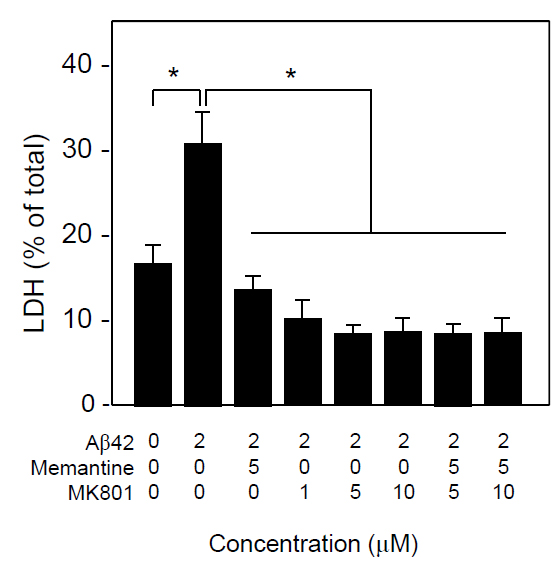
Because, memantine is known to be an uncompetitive NMDA receptor antagonist, we compared the neuroprotective effect of memantine and the NMDA channel blocker, MK801. As show in Figure 5, both memantine and MK-801 significantly inhibited Ab1-42-induced neuronal death, and there was no additive effect of memantine and MK-801 in terms of neuroprotection assayed by LDH release. These results suggest that memantine and MK-801 protect neurons in the same mechanism that is blocking NMDA channel.
2021 Copyright OAT. All rights reserv
Figure 5. Neuroprotective effects of memantine and MK801 in vitro. Neuronal cultures were treated with Aβ1-42 (3 μM) in the presence or absence of memantine and/or MK801 at the indicated concentrations. Forty-eight h later, the conditioned media was collected and the levels of released LDH were determined. Data are presented as the mean ± SD. N=3 per group. *P<0.005.
Discussion
Memantine is a drug prescribed to patients with AD for the amelioration of psychological symptoms and to slow the progression of this neurodegenerative disorder. It is thought that memantine exerts its beneficial effects on AD clinical symptomology and disease progression through NMDA receptor antagonism. However, several papers have reported that memantine can attenuate Aβ synthesis and Aβ burden, as well as ameliorate memory impairments in APP Tg mice, indicating that memantine may have other functions. Here, we sought to determine whether memantine had neuroprotective effects on Aβ-induced neuronal cell death in rat primary neuronal cultures. We found that memantine attenuated neuronal cell death induced by Aβ1-42 (3 μM), as determined by the LDH assay and Calcein AM and PI staining. We found that memantine attenuated neuronal death induced by Aβ1-42 (2 μM) and the neuroprotective effect of memantine may be due to its antagonism of the NMDA receptor.
The previous reports have shown that memantine, as a NMDA channel blocker, inhibits Aβ synthesis in vitro and in vivo, resulting in reduced APP, Aβ40, and Aβ42 levels as well as neuroprotection [19]. Memantine treatment also can reduce the total cortical levels of membrane-bound APP in both Tg and non-Tg mice, which may eventually lead to decreased Aβ[17]. Other lines of evidence show that memantine can improve memory impairments and the sAPPα/Aβ1-42 ratio[20,21], which are due to NMDA channel blockade[22,23]. Our present study has shown that in addition to Aβ synthesis, memantine can protect neurons from Aβ1-42-induced neurotoxicity in a dose-dependent manner. This effect was also seen with the competitive NMDA receptor blocker, MK801. There were no additive effects of memantine and MK801 in the attenuation of Aβ1-42-induced neurotoxicity, suggesting that the neuroprotective effect of memantine may be due to the blockade of the NMDA receptor. In agreement with our results, a previous study demonstrated that memantine can protect against Aβ-induced neuronal degeneration in vivo at therapeutically relevant doses [24]. It was also reported that memantine protects neurons against Aβ-induced neurotoxicity via prevention of tau phosphorylation [25], however, they did not show that this neuroprotection was via NMDA channel blockade.
Blockade of the NMDA receptor can inhibit glutamate-induced neuronal death. Previous reports have demonstrated that Aβ toxicity is associated with elevated glutamate levels and NMDA receptor hyperactivity. On the other hand, Aβ can also enhance the activation of extrasynaptic NMDA receptors by decreasing neuronal glutamate uptake and inducing glutamate spillover, resulting in neurotoxicity. The selective enhancement of synaptic activity by low doses of NMDA, or a reduction of extrasynaptic activity by memantine, can halt Aβ-induced neurotoxicity [26].In support of this, it has been shown that cellular damage in AD brains is prominent in areas of glutamatergic synaptic plasticity [27]. Several studies have suggested that Aβ-induced neurotoxicity is associated with glutamate excitotoxocity [28,29].
Interestingly, memantine attenuates LDH release in neuronal cultures in the absence of Aβ1-42. The formation of free radicals, which can induce cellular damage, occurs in neuronal cultures across time [30], suggesting that free radicals may exert their toxic effects via the NMDA receptor. Aβ and NMDA have been reported to induce reactive oxygen species in cortical neurons[31].Free radicals play a key role in neuronal damage in various neurodegenerative and vascular brain diseases. In addition, memantine is effective in preventing Aβ-induced short-term memory impairments [32], and can rescue both neuronal oxidative stress and transient memory impairments caused by high molecular weight oligomers. However, memantine did not rescue the persistent cognitive deficits induced by low molecular weight oligomers[33]. These data suggest that memantine may ameliorate symptoms in patients with other neurodegenerative and vascular diseases, in which ROS generation and/or the NMDA receptor play a role in the disease pathophysiology.The immunotherapy is now thought to be a promising therapy forAD, but it needs time to produce its effects. Thus the findings of this study with memantine as a protector against neuronal toxicity via NMDA induced by Aβ, could potentially and cooperatively ameliorate Aβ-induced brain damage and memory impairment in AD patients who have immunotherapy for AD.
Acknowledgments
This work was supported by a grant from the Ministry of Health, Labor and Welfare of Japan (Research on Dementia, Health and Labor Sciences Research Grants H23-005) (to M.M.).We have no conflict of interest to declare.
Conflict of interest
There is no conflict of interest to declare.
References
- Selkoe DJ (1994) Alzheimer's disease: a central role for amyloid. J Neuropathol Exp Neurol 53: 438-447. [Crossref]
- Glenner GG, Wong CW (1984) Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120: 885-890. [Crossref]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, et al. (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A 82: 4245-4249. [Crossref]
- Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, et al. (1993) beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci U S A 90, 10836-10840. [Crossref]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, et al. (1994) Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 13: 45-53. [Crossref]
- Mattson MP, Tomaselli KJ, Rydel RE (1993) Calcium-destabilizing and neurodegenerative effects of aggregated beta-amyloid peptide are attenuated by basic FGF. Brain Res 621: 35-49. [Crossref]
- Lorenzo A, Yankner BA (1994) Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci U S A 91: 12243-12247. [Crossref]
- Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB (1997) Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem 272: 22364-22372. [Crossref]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, et al. (1999) Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 19, 8876-8884. [Crossref]
- Zou K, Gong JS, Yanagisawa K, Michikawa M (2002) A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J Neurosci 22: 4833-4841. [Crossref]
- Parsons CG, Danysz W, Bartmann A, Spielmanns P, Frankiewicz T, et al. (1999) Amino-alkyl-cyclohexanes are novel uncompetitive NMDA receptor antagonists with strong voltage-dependency and fast blocking kinetics: in vitro and in vivo characterization. Neuropharmacology 38: 85-108. [Crossref]
- Danysz W, Parsons CG, Mobius HJ, Stoffler A, Quack G (2000) Neuroprotective and symptomatological action of memantine relevant for Alzheimer's disease--a unified glutamatergic hypothesis on the mechanism of action. Neurotox Res 2: 85-97. [Crossref]
- Martinez-Coria H, Green KN, Billings LM, Kitazawa M, Albrecht M, et al. (2010) Memantine improves cognition and reduces Alzheimer's-like neuropathology in transgenic mice. Am J Pathol 176: 870-880. [Crossref]
- Decker H, Lo KY, Unger SM, Ferreira ST, Silverman MA (2010) Amyloid-beta peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J Neurosci 30: 9166-9171. [Crossref]
- Scholtzova H, Wadghiri YZ, Douadi M, Sigurdsson EM, Li YS, et al. (2008) Memantine leads to behavioral improvement and amyloid reduction in Alzheimer's-disease-model transgenic mice shown as by micromagnetic resonance imaging. J Neurosci Res 86: 2784-2791. [Crossref]
- Alley GM, Bailey JA, Chen D, Ray B, Puli LK, et al. (2010) Memantine lowers amyloid-beta peptide levels in neuronal cultures and in APP/PS1 transgenic mice. J Neurosci Res 88: 143-154. [Crossref]
- Unger C, Svedberg MM, Yu WF, Hedberg MM, Nordberg A (2006) Effect of subchronic treatment of memantine, galantamine, and nicotine in the brain of Tg2576 (APPswe) transgenic mice. J Pharmacol Exp Ther 317: 30-36. [Crossref]
- Tucci P, Mhillaj E, Morgese MG, Colaianna M, Zotti M, et al. (2014) Memantine prevents memory consolidation failure induced by soluble beta amyloid in rats. Front Behav Neurosci 8: 332. [Crossref]
- Liu MY, Wang S, Yao WF, Zhang ZJ, Zhong X, et al. (2014) Memantine improves spatial learning and memory impairments by regulating NGF signaling in APP/PS1 transgenic mice. Neuroscience 273: 141-151. [Crossref]
- Spilman P, Descamps O, Gorostiza O, Peters-Libeu C, Poksay KS, et al. (2014) The multi-functional drug tropisetron binds APP and normalizes cognition in a murine Alzheimer's model. Brain Res 1551: 25-44. [Crossref]
- Nagakura A, Shitaka Y, Yarimizu J, Matsuoka N (2013) Characterization of cognitive deficits in a transgenic mouse model of Alzheimer's disease and effects of donepezil and memantine. Eur J Pharmacol 703: 53-61. [Crossref]
- Du H, Guo L, Wu X, Sosunov AA, McKhann GM, et al. (2014) Cyclophilin D deficiency rescues Aβ-impaired PKA/CREB signaling and alleviates synaptic degeneration. Biochim Biophys Acta 1842: 2517-2527. [Crossref]
- Xu Y, Cao DH, Wu GM, Hou XY (2014) Involvement of P38MAPK activation by NMDA receptors and non-NMDA receptors in amyloid-β peptide-induced neuronal loss in rat hippocampal CA1 and CA3 subfields. Neurosci Res 85: 51-57. [Crossref]
- Miguel-Hidalgo JJ, Alvarez XA, Cacabelos R, Quack G (2002) Neuroprotection by memantine against neurodegeneration induced by beta-amyloid(1-40). Brain Res 958: 210-221. [Crossref]
- Song MS, Rauw G, Baker GB, Kar S (2008) Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur J Neurosci 28: 1989-2002. [Crossref]
- Wang ZC, Zhao J, Li S (2013) Dysregulation of synaptic and extrasynaptic N-methyl-D-aspartate receptors induced by amyloid-β. Neurosci Bull 29: 752-760. [Crossref]
- Arendt T, Holzer M, Gärtner U, Brückner MK (1998) Aberrancies in signal transduction and cell cycle related events in Alzheimer's disease. J Neural Transm Suppl 54: 147-158. [Crossref]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, et al. (2007) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27: 2866-2875. [Crossref]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, et al. (2007) Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci 27: 796-807. [Crossref]
- Nagayasu Y, Morita SY, Hayashi H, Miura Y, Yokoyama K, et al. (2014) Increasing cellular level of phosphatidic acid enhances FGF-1 production in long term-cultured rat astrocytes. Brain Res 1563: 31-40. [Crossref]
- Shelat PB, Chalimoniuk M, Wang JH, Strosznajder JB, Lee JC, et al. (2008) Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J Neurochem 106: 45-55. [Crossref]
- Yamada K, Takayanagi M, Kamei H, Nagai T, Dohniwa M, et al. (2005) Effects of memantine and donepezil on amyloid beta-induced memory impairment in a delayed-matching to position task in rats. Behav Brain Res 162: 191-199. [Crossref]
- Figueiredo CP, Clarke JR, Ledo JH, Ribeiro FC, Costa CV, et al. (2013) Memantine rescues transient cognitive impairment caused by high-molecular-weight abeta oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J Neurosci 33: 9626-9634. [Crossref]