Computational analysis was carried out in the current study to analyze wild and mutant type (p.Cys283Asn, we identified in a previous study) of FBXO31 gene which has been identified to cause non‑syndromic autosomal recessive intellectual disability. Using bioinformatics tools, structure prediction, conservation analysis, pocket identification and docking interactions were executed. Results obtained demonstrated a clear difference in both forms of FBXO31. It was concluded that mutation produces a great change in normal structure, conformation and interaction site of FBXO31 protein which in turn causes a change in its interaction with other interacting proteins leading to change in protein function. Phylogenetic analysis showed that Human is closely related to Macaque and Chimpanzee. Synteny analysis indicated that highly conserved synteny of Human is with Chimpanzee and Mouse. Computational analysis has opened many insights into disease understanding in low cost and time and it will also provide a way towards accurate diagnosis and better therapeutic strategies.
ID, structure, MSA, phylogeny, synteny, docking, mutational analysis
Non-Syndromic Autosomal Recessive Mental Retardation or intellectual disability (NS-ARMR/ID) is the most important and serious heterogeneous neurodevelopmental disorder and a largely unresolved problem in genetic health care. It affects ~1 % of children worldwide It is diagnosed by a low intelligence quotient (IQ) i.e. 70 or below and deficits in adaptive behaviors such as delayed language, social skills or self-help skills. Also characterized by impaired capacity of learning and processing new or complex information, culminating in decreased adaptive behavior and cognitive functioning
Environmental factors, such as teratogens, infection, and neurological trauma are causes of ID. It has a strong genetic etiology as well as it includes a diverse range of genetic defects such as chromosomal aberrations, sub-microscopic copy number variations (CNVs), and DNA sequence mutations within genes, including genes located on the autosomes, X chromosome and the mitochondrial genome. Research into autosomal recessive forms of ID (ARID) has been largely missed, and only in recent years collective efforts have been made to identify ARID genes Recent estimates suggest that in Europe 13–24 % of ID cases are likely to be due to autosomal recessive causes, and may be the most common cause of ID in populations is consanguinity ID can be divided into two groups: non-syndromic ID is characterized as the clinical feature in patients only, while syndromic ID occurs in combination with one or more supplementary clinical features Forty genes have been reported to date for non-syndromic ARID
FBXO31 gene has been identified to cause Non‑Syndromic Autosomal Recessive Intellectual Disability FBXO31 performs function as a centrosomal E3 ubiquitin ligase, in association with SKP1 and Cullin-1. These are functional partners of FBXO31 which are involved in ubiquitination of proteins targeted for degradation. For neuronal morphogenesis and axonal identity, complex of the FBXO31, SKP1, and Cullin1 is very important. In dendrite growth and migration of neurons in developing cerebellar cortex FBXO31 plays a role The FBXO31 role has been well characterized as a regulator of cell-cycle and as a tumor suppressor gene in cancer research The FBXO31 importance as a centrosomal protein and a regulator in neuronal morphogenesis, where it controls the positioning and migration of axons and growth of dendrites, also gives an understanding to the function of this gene in the neuronal development.
In the current study, computational analysis has been carried out for normal FBXO31 gene along with a frame shifting, truncating mutation in the gene i.e. p.Cys283Asn (we identified in a previous study) in exon 7 of the gene. It causes a premature stop codon at position 362 resulting in a 177-amino acid truncation of FBXO31 protein. Mutation was associated with a form of autosomal recessive intellectual disability identified in family named ASMR72. The deletion and premature stop codon in the mutated form of FBXO31 appeared to correspond to reduce mRNA and protein levels. Moreover, in the study it was stated that the depletion of FBXO31 protein would likely affect the formation of axons and dendrites during neurogenesis The reason of analyzing this gene further using bioinformatics approach was to have its more exploration and deeper understanding. We also focused on mutation at every step during the analysis and analyzed both wild and mutant types of the gene. Using bioinformatics approach, structure prediction, conservation analysis, pocket identification and docking studies were executed for both wild and mutant FBXO31. Phylogenetic and synteny analysis was performed which gives an insight to evolutionary relationship that how species are related to each other in the form of ancestors/descendents and conservation, respectively among various selected ortholog species with reference to Human FBXO31.
A schematic representation of methodology used in the current study is given in Figure 1. Following is given description for every step.
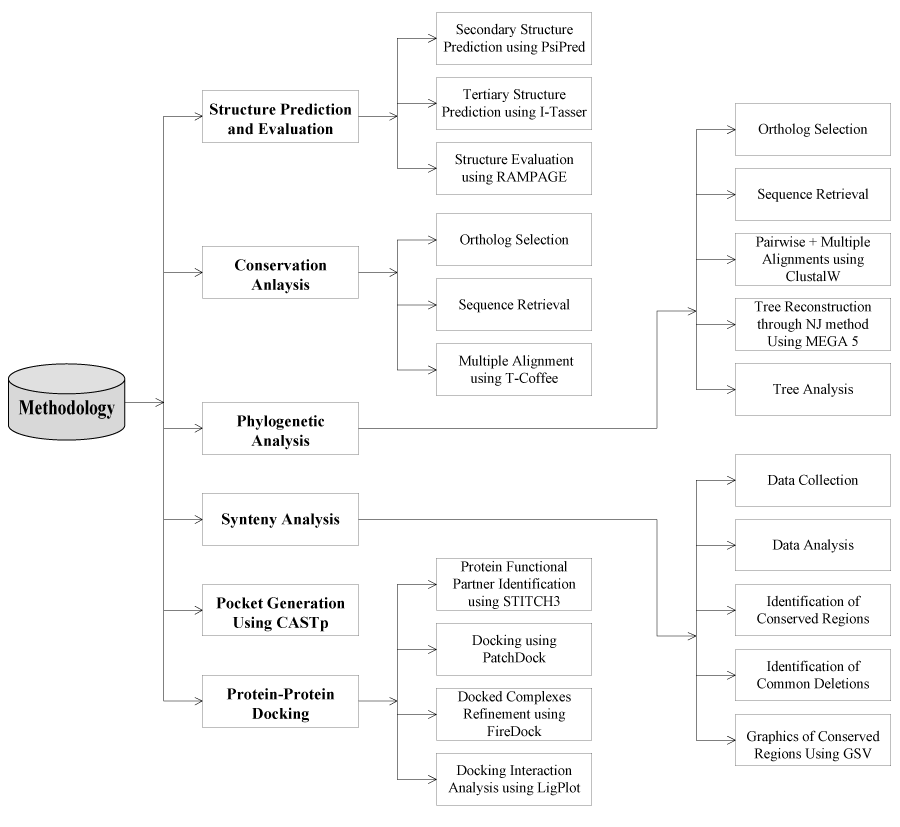
Figure 1. Design of the study of FBXO31.
Sequence acquisition
The FBXO31 gene comprises 9 exons, and encodes a 539-amino acid protein. Sequence was retrieved through OMIM database (MIM # 609102).
Structure prediction
Sequence of FBXO31 gene was retrieved through OMIM database (MIM # 609102). Three dimensional (3D) structure was built using threading approach via I-Tasser For evaluation of protein 3D models (wild + Mutated), RAMPAGE server was used.
Multiple sequence alignment
MSA (Multiple Sequence Alignment) was performed using T-Coffee (Tree-based Consistency Objective Function For alignment Evaluation) to show the consistency of sequence among various ortholog species of Human FBXO31 [14]. Thirteen ortholog species with reference to Human (Homo sapiens) have been considered for this study. These species include: Chimpanzee (Pan Troglodytes), Macaque (Macaca mulatta), Mouse (Mus musculus), Guinea Pig (Cavia porcellus), Megabat (Pteropus vampyrus), Dog (Canis familiaris), Hedgehog (Erinaceus Europaeus), Anole Lizard (Anolis carolinensis), Chicken (Gallus Gallus), Fugu (Takifugu rubripes), Zebrafish (Danio rerio), Opossum (Monodelphis domestica), Platypus (Ornithorhynchus Anatinus). Sequences of all ortholog species were collected through ensemble database.
Phylogeny
In the current study, MEGA5 [15] was used for phylogenetic tree reconstruction to estimate evolutionary relationship of Human FBXO31. Following is a brief description of methodology for this analysis.
Sequence retrieval
Thirteen ortholog species with reference to Human have been considered for phyloegenetic analysis. These species are same to those selected for conservation studies using T-Coffee in section of multiple sequence alignment. In order to select the sequences of ortholog species, sequence similarity of their sequences with Human gene sequence was analyzed through alignment using BLASTP (basic local alignment search tool) [16] against the protein database in order to choose closest putative orthologous protein sequences.
Phylogenetic tree reconstruction
ClustalW algorithm was used for pair-wise and multiple alignment. The algorithm calculates similarity percentage between sequences and generates an alignment file which is further used as input file during tree reconstruction. Statistical method in MEGA5 for tree reconstruction was Neighbor joining (NJ) [17]. Tree was generated based on this alignment file which was used as input for tree reconstruction. NJ shows a high performance as compare to rest methods in obtaining the correct tree but it’s more sensitive as compare to other methods and do not construct tree when evolutionary rate varies among the genes with high degree [18]. Bootstrap analysis was also used during tree reconstruction as phylogeney test which is a computer-based method that assigns accuracy measures to sample estimates [19]. It was done to test the reliability of resulting tree topology. Bootstrap values indicate cluster confidence and reliability. Tree topology is tested based on the bootstrap values which further validate the branching pattern. It is an accurate way to control and check stability of results obtained. In the current study bootstrap method uses 1000 bootstrap replications and assigns each branch a value ranging from 0 to 100 which gives an idea that how much a sequence is evolutionary closer to each other and also validates each branch. Only bootstrap values >70% were showed in tree. P-distance was chosen as a substitution model and our substitution type was amino acids.
Synteny analysis
Synteny analysis was performed using Ensemble synteny view in ensemble database [20] and the visual analysis of conserved regions was carried out using web-based genome synteny viewer GSV [21]. For this analysis only four ortholog species (Chimpanzee, Mouse, Chicken, and Pig) of Human have been considered.
Pocket identification
Pockets and voids present either on the surfaces or hidden in the interior sides on three-dimensional structures of proteins were identified using Computed Atlas of Surface Topography of proteins (CASTp)
Protein-protein docking studies
Protein interaction framework was analyzed through STITCH3 database [24]. Protein-protein docking analysis was carried out using PatchDock server [25, 26]. Total 100 runs were carried out to generate best docking complex. The first 10 docked complexes were retrieved and then subjected to FireDock server [27, 28] for further analysis (i.e. refinement and ranking of interactions). Docked complexes of protein and ligands were visualized using ViewerLite software version 5.0. After docking, 2D (two dimensional) representations of protein-ligand complexes and their interactions were generated and analyzed using LIGPLOT program [29]. For Protein ligand it has an option of DimPlot to study intermolecular interactions. Standard input files of Protein Data Bank (PDB) were used for this purpose by the program. This program facilitates rapid inspection of many complexes. PostScript file (in colored or black-and-white form) was generated as an output. Intermolecular interactions in a simple and informative form were given in this output file and their strengths (which includes hydrogen bonds, hydrophobic interactions and atom accessibilities) as well.
Structure prediction and evaluation
According to Psipred results (Figure 2) for protein secondary structure prediction it has been predicted that in normal FBXO31 structure there are 9 helixes, 15 strands and 24 coils. In mutated FBXO31 number of helixes, strands and coils gets affected and becomes 8, 6, and 15, respectively. Amino acid Cysteine at position 283 is involved in making coil structure, hence after its substitution by asparagine and frameshift, size of coil and number of structure features gets affected.
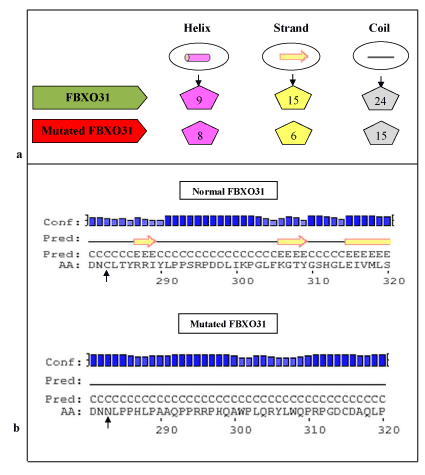
Figure 2. Psipred Results for Normal and Mutated FBXO31: a) Comparison of Predicted Number of Structure Features, b) Mutation Area (substitution of C at position 283 with N highlighted with arrowhead.
By folding of alpha helices and beta sheets into compact globule form, third level of proteins are formed which is considered as tertiary (3D) structures of proteins. To predict 3D structure of normal and mutated FBXO31, threading approach via I-TASSER was used. Structures shown in Figure 3 containing helices, beta sheets, and coils. Evaluation of these predicted structures has shown residues in favored, allowed and outlier regions. Maximum percentage of residues lies in the favored region which shows that predicted structures are reliable. In case of normal structure 440 (81.9%), 64 (11.9%), 33 (6.1%) and in case of mutant type, 288 (80.0%), 51 (14.2%), and 21 (5.8%) residues percentages lies in the favored, allowed and outlier regions, respectively. This analysis also shows difference in both structures (wild and mutated) through difference in the number of residues in favored, allowed and outlier regions. Mutation not only affects structure and conformation but also function of protein, leading to the diseased state.
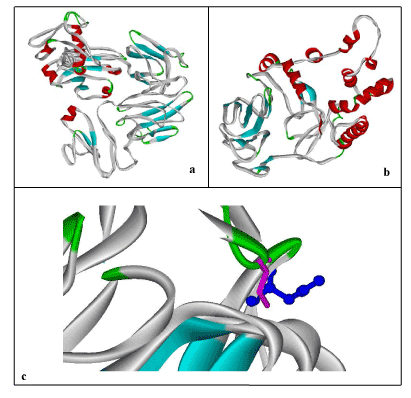
Figure 3. 3D-Structure Prediction by I-Tasser: a) Human FBXO31, b) Mutated FBXO31, c) Superimposed Normal and Mutated Structures to highlight mutation; Cystine at Position 283 (Display Style: Stick, Color: Pink) replaced by Asparagine (Display Style: Ball and Stick, Color: Blue). Structures have been shown in Solid Ribbon display style via Viewerlite v5.0.
Conservation analysis
Multiple sequence alignment was carried out using T-Coffee to find the conservation of amino acids Cysteine (C) at positions 283 which after mutation was replaced by Asparagine (N). Results showed 100% conservation of this amino acid among thirteen ortholog species (Figure 4). Conservation of Cysteine indicates its importance in the sequence.

Figure 4. T-Coffee Results: Multiple Sequence Alignment for Human FBXO31 gene. Conservation of amino acids C at position 283 among various ortholog species is indicated by arrow.
Phylogenetic analysis
Neighbor joining tree for FBXO31 is shown in Figure 5. According to tree, Human is making cluster with Macaque with bootstrap value of 75. With a good bootstrap value of 99, Chimpanzee is close to the cluster of Human and Macaque. This also indicates that Human is closely related to Chimpanzee as compare to other species. Bootstrap values only greater than 70 are mentioned in tree. Other clusters with high bootstrap values include Platypus and Oppossum, Anoli Lizard and Chicken, Platypus/Oppossum/Anoli Lizard/Chicken and Dog/Megabat/Hedgehog/Guinea Pig/Mouse/Chimpanzee/Macaque with their respective bootstrap values of 99, 90, and 100. High bootstrap values of the clusters show their reliability. Tree is reconciling species divergence time. Fugu and Zebrafish are as outgroup in this tree. Evolutionary time for the tree is 0.01.
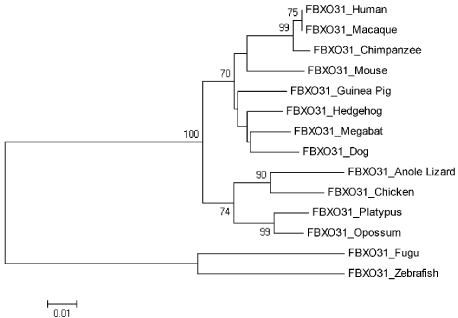
Figure 5. Neighbor Joining (NJ) Tree for Human FBXO31 using MEGA5, Numbers on Branches Represent Bootstrap Values Based on 1000 Replications.
Genome synteny analysis
In order to find out the genomic elements that are functionally conserved, we find out set of genomic features (genes or loci) that are conserved, in the same relative ordering on a set of homologous chromosomes (of human and its four orthologs). We studied conservation of human 15 genes (both upstream and downstream of FBXO31) with genes of its four orthologs. Data collected from ensembl syntenyview in ensembl database.
Four orthologs Chimpanzee (Pan troglodytes), Mouse (Mus musculus), Chicken (Gallus gallus) and Pig (Sus scrofa) were selected for this analysis. These studies clearly demonstrate the presence and absence of conserved synteny between Human and these orthologs. Using genome synteny viewer (GSV) web server also conserverd regions were generated as shown in Figure 6. Graphical representations generated by GSV facilitated the quick visualization of conserved regions in the form of colored blocks. Ruler in the figure is indicating positions of these conserverd regions. Number of colored blocks shows majority of the portion is conserved between orthologs and Human. Majority of the portion is conserved among two orthologs i.e. Chimpanzee and Mouse in relevance to Human with only 5 deletions as per our analysis. Less conservation was observered for Chicken and Pig with 9 and 15 deletions, respectively. In Table 2 there are given changes in the form of deletions which lead evolution of these organisms. Common deletions in four orthologs in relevance to Human FBXO31 are four i.e. CTC-786C10.1, RP11-680G10.1, AC010536.1, FLJ00104 (Table 1). FBXO31 is present in all four ortholog species which shows its importance in these species.
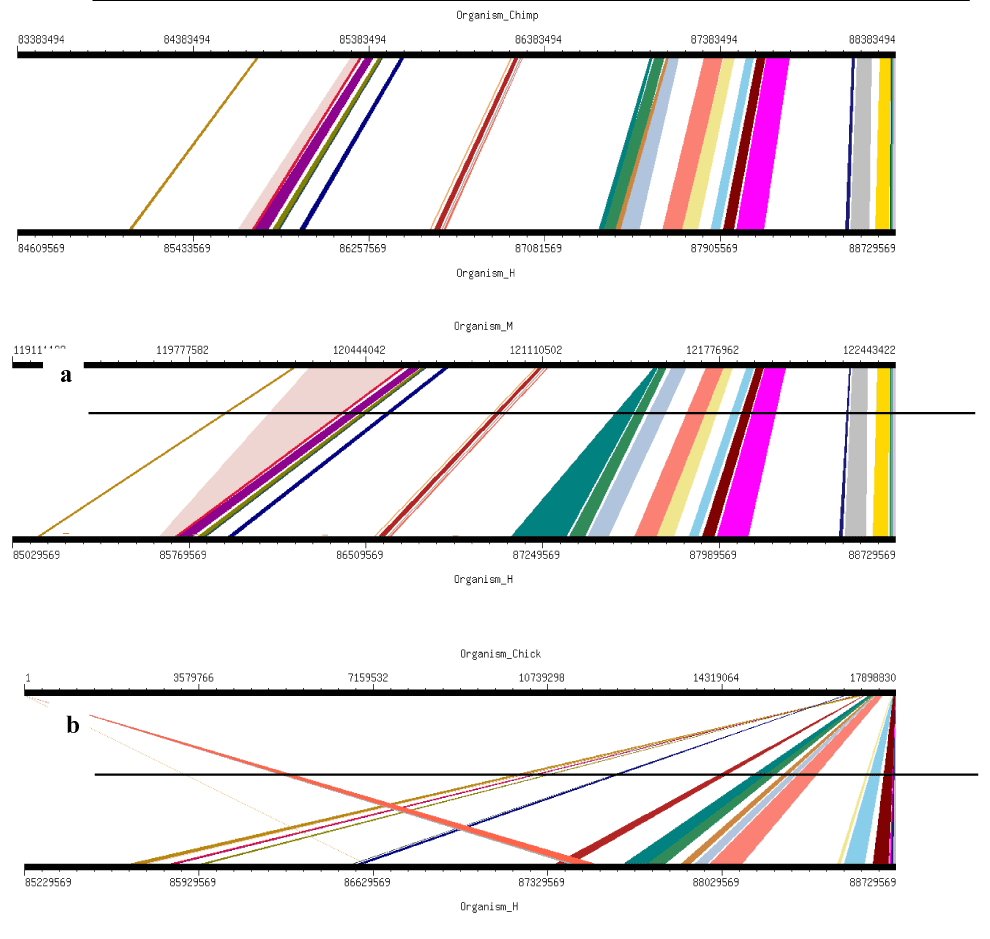
Figure 6. Results of GSV for Human FBXO31 showing Conserved Regions: a) Organism_Chimp (Chimpanzee) vs Organism_H (Human); b) Organism_M (Mouse) vs Organism_H (Human); c) Organism_Chick (Chicken) vs Organism_H (Human); d) Organism_P (Pig) vs Organism_H (Human)
Table 1. Observed Total and Common Deletions in Four Orthologs with Reference to Human FBXO31
|
Organism
|
Observed No. of Deletions
|
Deleted Genes
|
|
Chimpanzee
|
5
|
CTC-786C10.1, RP11-680G10.1, C16orf95, AC010536.1, FLJ00104
|
|
Mouse
|
5
|
CTC-786C10.1, RP11-680G10.1, AC010536.1, FLJ00104, RP11-178L8.4
|
|
Chicken
|
9
|
CTC-786C10.1, RP11-680G10.1, AC010536.1, FLJ00104, RP11-178L8.4, FAM92B, C16orf74, FOXL1, C16orf95
|
|
Pig
|
15
|
CTC-786C10.1, RP11-680G10.1, AC010536.1, FLJ00104, RP11-178L8.4, FOXF1, FOXC2, SLC7A5, CA5A, ZNF469, ZFPM1, ZC3H18, IL17C, CYBA, MVD
|
Table 2. Receptor (FBXO31) and Ligand (CUL1) Residues Involved in Interactions
|
Receptor-Ligand
|
Hydrogen Bond Interactions
|
Hydrophobic Interactions
|
Figure
|
|
|
Ligand Residues
|
Receptor Residues
|
Ligand Residues
|
Receptor Residues
|
|
|
FBXO31-CUL1
|
Val20,Ala38, Glu44,Tyr119, Thr120,Trp123, Ser130,Asn134, Arg146,Gly20, Ser207,Glu210
|
Val452,Ser454, Asp458,Gln360, Arg455,Asn456, Tyr459,Glu482, Pro485
|
Arg34,Ser36,Met37,Lys39,Ser40, Arg41,Tyr42,Met43,Glu124, Asp125, Tyr126, Arg127, Phe128, Ser129,Lys131,Val132,Leu133, Gly135, Ile136, Cys137, Ala138, Tyr139, Asn141, Arg142, His143, Val145, Arg147, Glu148, Cys149, Lys154, Gly155, Ile156, Tyr157, Glu158, Ile159, Tyr160, Ser161, Leu162, Ala163, Leu164, Val165, Trp167, Arg168, Val205, Gln206, Tyr208, Val209, Leu211, Gly212, Leu213
|
Glu358, Asn359, Arg408, Ala409, Glu422, Glu426, Gly428, Asp429, Ala430, Val431, Ala432, Gln444, Pro445, Phe446, Val447, Leu448, Pro449, Val450, Gly451, Ser453, Glu457, Pro460, Arg461,Thr462, Cys463, Ile473, Ala474, Gly475, His476, Gly477, Phe478,Thr479, Ser480, Pro481, Arg483, Thr484, Gly486, Val487, Phe488, Ile489
|
9(a)
|
|
Mutated FBXO31-CUL1
|
Asp109
|
Arg64
|
Asp18, Trp21, Asp22, Lys39, Ser40, Glu44, Asp106, Glu108, Leu110
|
Asp36, Thr37, Asp38, Pro39, Glu41, Pro62, Pro63, Cys65, Leu90, Val93, Ala353, Arg354
|
9(b)
|
Active site analysis
Proteins exhibit specific shapes due to their secondary and tertiary structure. Binding site, pocket or active site is region where specific molecules bind, called ligands. It is base of lock and key model. Interaction of molecules at active site depicts chemical change / reaction. In proteins, active site is normally hydrophobic pocket that includes side chain atoms. Indicating molecules that bind to target protein is helpful for 3D structure and important as well for drug designing. Moreover these pockets involve specific amino acids hence their prediction is also useful for mutational analysis. A total of 93 pockets generated through CASTp for FBXO31 protein out of which pocket 82 contains amino acids Cysteine (C) at positions 283 as shown in Figure 7. In this figure pocket number 82 has been highlighted in pink color. Results show importance of this amino acid and also indicate that mutation is not only changing structure of protein but it is affecting active sites as well. Due to deletion of amino acid, structure, conformation, active sites and finally function of protein changes leading to the diseased state from normal state. Total number of pockets in normal FBXO31 structure is 93. In mutated FBXO31 structure total number of pockets becomes 54 due to replacement of amino acid C by N and truncation of protein.
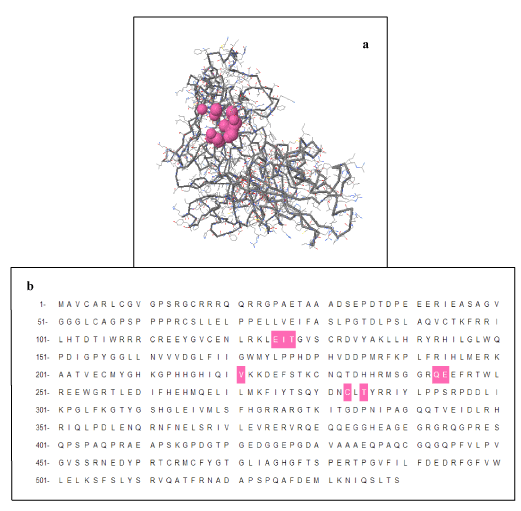
Figure 7. CASTp Results: Pocket Number 82 Highlighted in pink color contains Amino Acid C at positions 283: a) Structure Representation, b) Sequence Representation.
Docking interaction studies
To obtain some material evidence of the assumption that proteins involved in FBXO31 belong to a functional pathway and interact at some level with associated proteins, Stitch3 server was used. Predicted functional partners of FBXO31 gene is shown in Figure 7. Protein with maximum interaction score (0.965) with FBXO31 was selected for further analysis and considered as protein ligand i.e. CUL1. Both normal and mutated structures of FBXO31 were docked with this protein ligand to study the docking process as well as effect of mutation. Figure 8 (a) shows docking complex of FBXO31 receptor and CUL1 ligand while Figure 9 (b) demonstrates docking complex of Mutant FBXO31 receptor and CUL1 ligand.
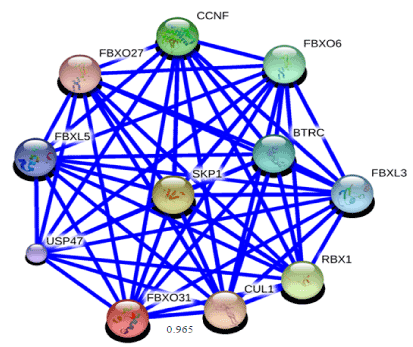
Figure 8. Functional partners of FBXO31 predicted using STITCH3 server. Highest interaction score of FBXO31 is with CUL1 i.e. 0.965
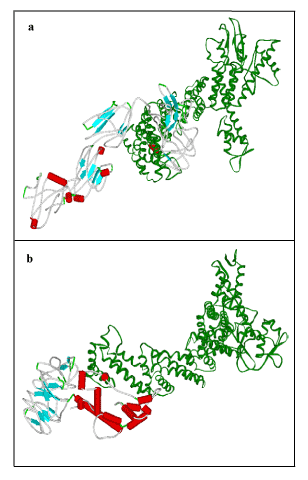
Figure 9. Visualization of Docking Results using Viewer lite: a) FBXO31 with CUL1, b) Mutated FBXO31 with CUL1. Receptor: Color by secondary type and Schematic display style, Ligand: Green Color, Solid Ribbon display style.
Wild structure of FBXO31 changes upon mutation and converts into mutated structure. Change in the structure also causes alteration in the conformation and hence interaction sites of protein (i.e. active sites). Active site is the location on receptor protein with which ligand interacts and binds. As active site changes when a wild type structure gets mutated hence ligand interaction also changes along with. Table 3 contains docking results of receptor/ligand residues involved in the interaction for FBXO31 (normal and mutated) with ligand CUL1. Specific residues of ligand and receptor are involved in docking interaction. These ligand/receptor residues dock with the help of non-covalent interactions, for example hydrogen bonding and hydrophobic interactions. Residues of both receptor and ligand that were involved in hydrogen bonding and hydrophobic interaction as a result of both docking interactions are given in Table 2. These results show that there is great difference in the interaction sites of docked complexes of FBXO31 (normal and mutated) with ligand CUL1. Difference in the interaction sites can be estimated through the amino acids which are involved in the hydrogen bonding and hydrophobic interactions between ligand and binding site of receptor protein. Dimplot results for the docking interactions of FBXO31 (normal and mutated) with ligand CUL1 are shown in Figure 10. It also evaluates our docking results that there is a difference in the docking interaction of normal/wild type structure of FBXO31 and its mutated form with CUL1 ligand. Substitution of amino acids Cysteine at position 283 by Asparagine and frameshift has caused a great change in size, structure, and conformation and interaction site of protein. This change in structure, conformation and interaction site causes a big change in function of the protein leading to the disease state.
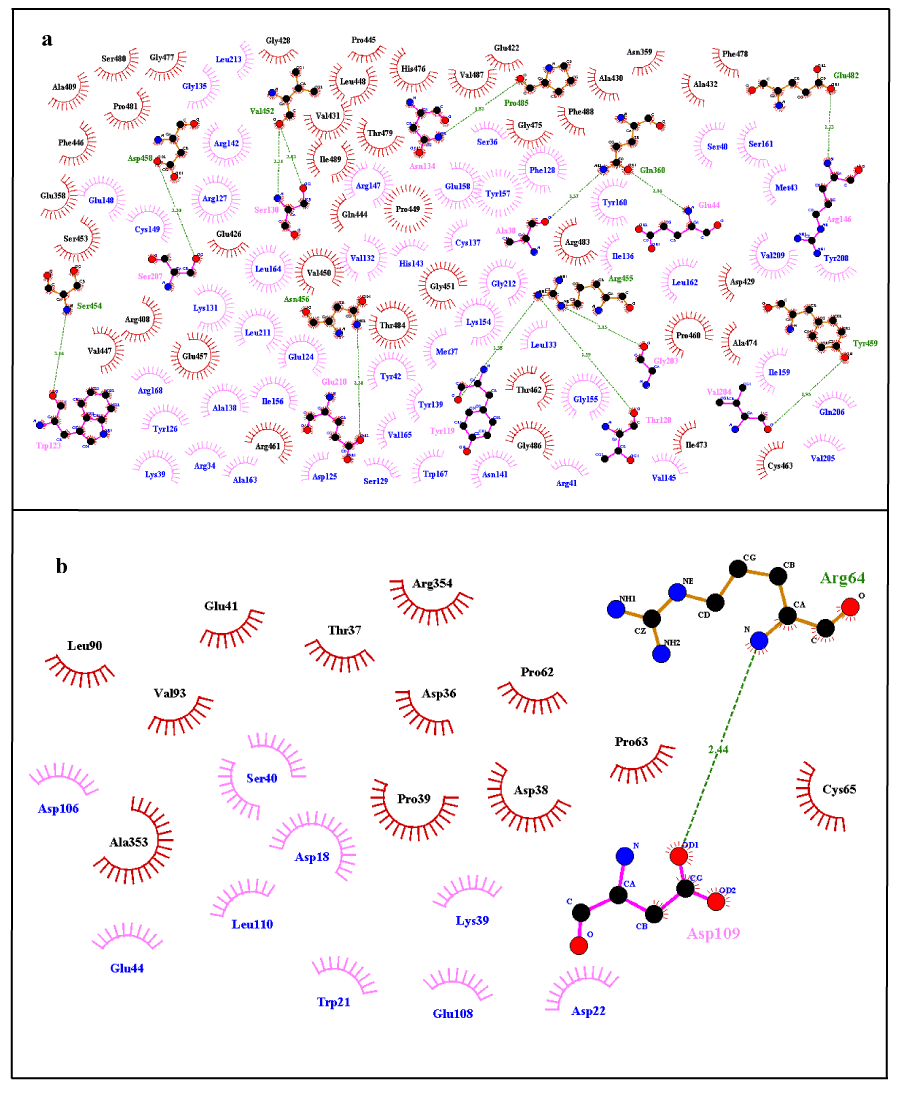
Figure 10. Dimplot Results for Docking Interactions; a) Normal FBXO31 with CUL1, b) Mutated FBXO31 with CUL1. Ligand Residues Involved in Hydrophobic Interactions are shown in blue color and represented by pink spoked arcs ( ). Receptor Residues Involved in Hydrophobic Interactions are shown in black color and represented by brick red spoked arcs ( ). Green dotted lines ( ) show Hydrogen Bonding. Receptor residues involved in H-Bonding are shown in Olive Green Color. Ligand residues involved in H-Bonding are shown in pink color.
Computational analys2021 Copyright OAT. All rights reservdy to analyze wild and mutant types of FBXO31 gene. Using bioinformatics approach, structure prediction, conservation analysis, phylogeny and synteny analysis, pocket identification and docking studies were performed for both wild and mutant FBXO31. Psipred results for protein secondary structure prediction determined that mutation has affected number of structure features of normal FBXO31 i.e. helixes, strands and coils. 9 helixes, 15 strands and 24 coils of normal FBXO31 structure became 8, 6, and 15, respectively after mutation. Amino acid Cysteine at position 283 was involved in making coil structure. Consequence of this amino acid substitution was effect on the coil size and number of structure features as size of protein was reduced due to frame shift. Three dimensional structures for wild and mutated forms of FBXO31 protein was predicted and evaluated using I-Tasser and RAMPAGE, respectively. Reliability of predicted structures was obtained on the basis of maximum percentage of residues which were lying in the favored region. These results also indicated difference in normal and mutated structure on the basis of number of residues in favored, allowed and outlier regions.
T-Coffee results showed 100% conservation of amino acid C (deleted as a result of mutation) among thirteen ortholog species and indicated its importance. Phylogenetic analysis showed that Human is closely related to Macaque and Chimpanzee. From our present results, we hypothesized that due to having closest relationship, it is possible that mutations can affect Macaque and Chimpanzee (closest Human relative according to our results) likewise as these affect Human and can lead to disease state. It shows that apart from sequence similarity, function of gene in closely related species is also same and this function disrupts as a result of mutation and hence leads to the diseased state. Synteny analysis showed that majority of the portion is conserved among two orthologs i.e. Chimpanzee and Mouse in relevance to Human with only 5 deletions. Common deletions in four orthologs in relevance to Human FBXO31 were CTC-786C10.1, RP11-680G10.1, AC010536.1, and FLJ00104. Importance of FBXO31 can be estimated through the fact that it was present in all four ortholog species.
CASTp results showed that for FBXO31 protein, out of 93 total pockets, pocket number 82 contains amino acids C at position 283. In mutated FBXO31 structure total number of pockets became 54. Results showed importance of this amino acid and also indicated that mutation is not only changing structure of protein but it is affecting active sites as well. Due to mutation, a change in protein length, structure, conformation and active sites occurred. This affects protein function leading to the diseased state.
According to stitch database, protein with maximum interaction score of 0.965 with FBXO31 was CUL1. It was considered as protein ligand for docking analysis. Docking results showed great difference in the interaction sites of docked complexes of FBXO31 (normal and mutated) with ligand. Dimplot results showed a difference in the interaction sites on the basis of difference in amino acids which were involved in the hydrogen bonding and hydrophobic interactions between ligand and binding site of receptor protein. In docking interaction analysis of mutated FBXO31 with CUL1 it was not present due to deletion event. Substitution of amino acids Cysteine at position 283 by Asparagine and frame shift has caused a great change in size, structure, and conformation and interaction site of protein. This in turn causes a change in its interaction with other interacting proteins leading to change in protein function. This change in structure, function and interaction produces disease state.
In the current study, apart from analysis of wild and mutant types of gene, information about FBXO31 normal structure, conformation, interaction site and residues actively involved in interaction during protein-protein docking can be used to develop drug therapy for affected patients. Computational analysis has opened many insights into disease understanding in cost and time effective way and it will also provide a path towards accurate diagnosis and better therapeutic strategies.
We are thankful to Department of Bioinformatics and Biotechnology, International Islamic University, Islamabad for providing us a working platform. We are also thankful for the online available tools, databases and softwares which helped in making this research come to a conclusion.
- Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S (2011) Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev Disabil 32: 419-436. [Crossref]
- Ellison JW, Rosenfeld JA, Shaffer LG (2013) Genetic basis of intellectual disability. Annu Rev Med 64: 441-450. [Crossref]
- Leonard H, Wen X (2002) The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment Retard Dev Disabil Res Rev 8: 117-134. [Crossref]
- Musante L, Ropers HH2 (2014) Genetics of recessive cognitive disorders. Trends Genet 30: 32-39. [Crossref]
- Kaufman L, Ayub M, Vincent JB (2010) The genetic basis of non-syndromic intellectual disability: a review. J Neurodev Disord 2: 182-209. [Crossref]
- Mir A, Sritharan K, Mittal K, Vasli N, Araujo C, et al. (2014) Truncation of the E3 ubiquitin ligase component FBXO31 causes non syndromic autosomal recessive intellectual disability in a Pakistani family. Hum Gene 1-10. [Crossref]
- Kumar R, Neilsen PM, Crawford J, McKirdy R, Lee J, et al. (2005) FBXO31 is the chromosome 16q24.3 senescence gene, a candidate breast tumor suppressor, and a component of an SCF complex. Cancer Res 65: 11304-11313. [Crossref]
- Huang HL, Zheng WL, Zhao R, Zhang B, Ma WL (2010) FBXO31 is down-regulated and may function as a tumor suppressor in hepatocellular carcinoma. Oncol Rep 24: 715-720. [Crossref]
- Kanie T, Onoyama I, Matsumoto A, Yamada M, Nakatsumi H, et al. (2012) Genetic reevaluation of the role of F-box proteins in cyclin D1 degradation. Mol Cell Biol 32: 590-605. [Crossref]
- McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics 16: 404-405. [Crossref]
- Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nature Protocols 5: 725. [Crossref]
- Zhang Y I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9: 40. [Crossref]
- Lovell SC, Davis IW, Arendall WB 3rd, de Bakker PI, Word JM, et al. (2002) Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Protein Struct Funct Genet 50: 437-450. [Crossref]
- Notredame C, Higgins GD, Heringa J (2000) T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302: 205–217. [Crossref]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol 28: 2731–2739. [Crossref]
- Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, et al. (2008) NCBI BLAST: a better web interface. Nucleic Acids Res 36: W5-9. [Crossref]
- Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4: 406-425. [Crossref]
- Saitou N, Imanishi T (1989) Relative Efficiencies of the Fitch-Margoliash, Maximum- Parsimony, Maximum-Likelihood, Minimum-Evolution, and Neighbor-joining Methods of Phylogenetic Tree Construction in Obtaining the Correct Tree. Mol Biol Evol 6: 514-525.
- Efron B, Tibshirani JR (1993) An introduction to the bootstrap, Chapman & Hall/CRC. 1-430.
- Hubbard T, Barker D, Birney E, Cameron G, Chen Y, et al. (2002) The Ensembl genome database project. Nucleic Acids Res 30: 38-41. [Crossref]
- Revanna KV, Chiu CC, Bierschank E, Dong Q (2011) GSV: a web-based genome synteny viewer for customized data. BMC Bioinformatics 12: 316. [Crossref]
- Binkowski TA, Naghibzadeh S, Liang J (2003) CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res 31: 3352-3355. [Crossref]
- Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, et al. (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucl Acids Res 34: W116-W118. [Crossref]
- Kuhn M, Szklarczyk D, Franceschini A, von Mering C, Jensen LJ, et al. (2012) STITCH 3: zooming in on protein-chemical interactions. Nucleic Acids Res 40: D876-880. [Crossref]
- Duhovny D, Nussinov R, Wolfson HJ. (2002) Efficient Unbound Docking of Rigid Molecules. LNCS 2452: 185-200.
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ (2005) PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res 33: W363-367. [Crossref]
- Andrusier N, Nussinov R, Wolfson HJ (2007) FireDock: fast interaction refinement in molecular docking. Proteins 69: 139-159. [Crossref]
- Mashiach E, Schneidman-Duhovny D, Andrusier N, Nussinov R, Wolfson HJ (2008) FireDock: a web server for fast interaction refinement in molecular docking. Nucleic Acids Res 36: W229-232. [Crossref]
- Wallace CA, Laskowski AR, Thornton MJ (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng Des Sel 8: 127-134. [Crossref]