Abstract
We have studied a 60 year old male of Chinese descent who presented with combined deficiency of the anticoagulant proteins C and S. Molecular genetic analysis of the patient’s PROC gene revealed a novel heterozygous mutation that resulted in a V221G mutation. Two mutations were identified in the patient’s PROS1 gene: (1) an E67E mutation that has been reported previously and is associated with severe protein S deficiency, and (2) a novel mutation in the 5’ flanking sequence that changes a putative binding site for the transcription factor Sp 1. Together, these mutations are consistent with the combined protein C and S deficiency symptoms presented by the patient.
Key words
thrombosis, autosomal dominance, anti-coagulation, mutation, promoter
Abbreviations
PC: protein C, PS: protein S
Introduction
Proteins C and S (PC and PS) are vitamin K-dependent plasma glycoproteins that exert their anticoagulant actions through the degradation of clotting factors VIIIa and Va. After activation by the thrombin-thrombomodulin complex, activated protein C (APC) acts as a specific protease with PS acting as a non-enzymatic cofactor [1,2]. The human gene encoding PC (PROC) is located on the long arm of chromosome 2 (2q13-q14) and contains 9 exons that code for 461 amino acid residues [3,4]. The human PS gene (PROS1) resides on chromosome 3 (3p11.1-q11.2) and contains 15 exons coding for 636 amino acid residues [5-7]. The human genome also contains a pseudogene for protein S (PROSP or PROS2) also located on chromosome 3 (3p21-cen) [5-7].
Hereditary deficiencies of either PC or PS are rare autosomal dominant disorders in which patients have functional levels of the respective proteins that are typically half those of normal controls, resulting in increased propensity toward thromboembolic disease [2,8,9]. Like other inherited hemostatic disorders, deficiencies of PC or PS are the result of heterogeneous deletions and mutations that affect protein secretion, stability and/or function. Although over 150 PC [8] and 200 PS [10] mutations have been identified, mutations in the PS gene can only be identified in about 50% of cases with PS deficiency leading to the suggestion that causative genetic defects may be located close to the PS gene in these cases [11]. Combined PC and PS deficiency is a very rare disorder and few cases have been described in the literature [12]. Combined PC and PS deficiency is associated with increased risk of thrombosis [12-17] and stroke [18]. The molecular basis of the combined deficiency was determined in one family and revealed independently-segregating mutations in the PROS and PROC genes [13].
In this paper, we describe a patient who has combined deficiency of both PC and PS. Molecular genetic analysis of the patient’s genomic DNA revealed two previously unreported heterozygous mutations in the PC and PS genes. These genetic mutations are consistent with the low levels of proteins C and S that were observed in this patient.
Materials and methods
Case summary
The propositus is a 60 year old male of Chinese ancestry with no history of thrombosis. The propositus’ brother developed an unprovoked deep vein thrombosis at age 47 and was found to be deficient in both PC and PS. A thrombophilia work up on the propositus revealed a PC proteolytic activity level of 0.33 (normal > 0.50), a PTT-based PS activity level of 0.45 (normal > 0.60), and an ELISA-based PS free antigen level of 0.50 (normal > 0.50). These results were nearly identical to those of his brother and are consistent with combined heterozygous deficiencies of both anticoagulant proteins. In addition, the propositus' sister was found to be deficient in PC but had completely normal PS levels and no history of thromboembolic disease. Unfortunately, the patient's parents and siblings were unavailable for comparative molecular analysis.
Molecular genetic analysis
To determine the molecular basis for the PC and PS deficiencies in the propositus, informed consent was obtained and DNA was purified from citrated blood using the QIAamp DNA Isolation Kit (Qiagen, Mississauga, ON, Canada). Polymerase Chain Reaction (PCR) amplifications were performed using oligonucleotide primers flanking all exons and adjacent intron sequences of the PROC and PROS1 genes (primer details are available from the corresponding author). The PCR products were purified with the QIAquick PCR Purification Kit (Qiagen), and their nucleotide sequences were determined using the BigDye Terminator v3.0 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). Reaction products were analyzed on an Applied Biosystems 3700 DNA Analyzer located at the British Columbia Centre for Excellence in HIV/AIDS at St. Paul’s Hospital (Vancouver, BC). DNA sequences were determined on both strands of DNA and analyzed using Chromas 2.33 software (Technelysium Pty Ltd, Tewantin, Australia) and the basic local alignment search tool (BLAST) from the National Center for Biotechnology Information (NCBI). Assignment of nucleotide numbering was according to NCBI accession numbers NM_000312.2 for PROC and NM_000313.1 for PROS1.
Results
Both the PROC and PROS1 genes were amplified using specific PCR primers, and the resulting DNA fragments were subjected to DNA sequence analysis. A heterozygous missense mutation (PROC c.882 T>G) was identified in exon 8 of the PROC gene (Figure 1A). This mutation results in a valine to glycine substitution of residue 221 in the proteolytic domain of PC. This mutation has not been reported previously and is not reported in the database of PROC mutations (http://www.itb.cnr.it/procmd).
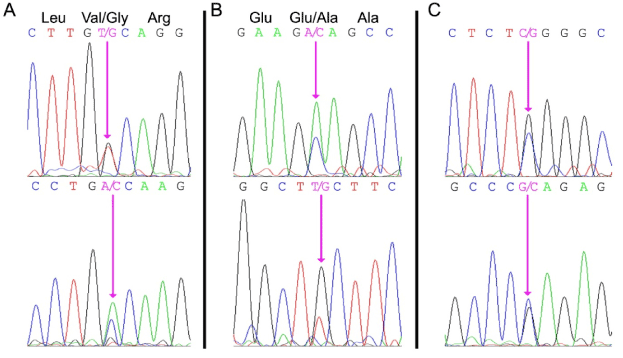
Figure 1. DNA sequence analysis of the patient’s genomic DNA.
Panel A: PROC exon 8 region illustrating heterozygosity for the PROC c.882 T>G missense mutation resulting in a ValàGly substitution at residue 221.
Panel B: PROS1 exon 2 region illustrating heterozygosity for the PROS1 c.200 A>C mutation resulting in a GluàAla substitution at residue 67.
Panel C: PROS1 promoter region illustrating heterozygosity for the PROS1 g.-190 C>G mutation within a highly-conserved proposed Sp1 transcription factor binding site.
The forward sequence is shown in the top panel and the reverse sequence is shown in the bottom panel. The mutated nucleotides are indicated by the pink arrows.
DNA sequence analysis of the patient’s PROS1 gene revealed two heterozygous mutations suggesting that the PS deficiency was not caused by a gross deletion of one of the alleles [11]. An exon mutation (PROS1 c.200 A>C) was identified in exon 2 of the patient's PROS1 gene (Figure 1B). This mutation has been reported previously [19] and results in the substitution of Glu67 to Ala. Glu67 is required for vitamin K-dependent proteins to bind Ca2+ and bind to phospholipid membranes [20]. Glu67 also appears to be important for protein S secretion from the liver into blood [19]. A second mutation was identified in the 5’ flanking sequence of the PROS1 gene (Figure 1B), and represented a heterozygous CàG nucleotide substitution at a position 190 bp upstream of the translational start site (Figure 1C). This substitution (PROS1 g.-190 C>G) is present within a proposed Sp1 transcription factor binding site that is highly conserved among mammals [21].
Discussion
In this study, we have identified two novel mutations and a previously reported mutation in the PROC and PROS1 genes of a patient with combined deficiency of PC and PS. Many different PROC mutations have been described in the literature and are involved in the various functional domains of this vitamin K-dependent zymogen (http://www.itb.cnr.it/procmd). The V221G mutation found in the current study has not been described previously in the PROC gene. However, there is a report of the mutation of the equivalent residue in Factor IX (Val231) being mutated to Phe and resulting in a severe hemophilia B with < 1% of normal factor IX activity [22]. Unfortunately, no biochemical analysis of the resulting V231F factor IX has been reported so the effect of the mutation on factor IX stability or enzymatic activity remains unknown. The patient’s low APC activity is consistent with the mutation of the equivalent valine residue and probably reflects the importance of this residue to the stability or activity of the resulting mutated APC.
Two mutations were identified in the patient’s PROS1 gene. Because the patient’s parents and siblings are unavailable for molecular genetic analysis, we are unable to determine if the two PROS1 gene mutations occur on a single allele or are distributed on both alleles. However, the G67A mutation alone is sufficient to result in the PS deficiency phenotype presented by the propositus [19]. Although another mutation has been identified in the PROS1 promoter region in a PS deficient patient [21], the current mutation has not been previously reported in the ISTH PROS1 mutation database [10]. Further studies would be required to confirm that the mutated sequence reduces the promoter activity in the mutant PROS1 gene but a reduction in PROS1 gene transcription with a resulting reduction in PS mRNA and plasma protein is consistent with the patient’s hematological presentation.
In conclusion, we have identified two novel genetic defects that are consistent with the causative molecular deficiencies of anticoagulant proteins C and S in a patient with heritable thrombophilia. To our knowledge, this represents only the second study involving the determination of the molecular basis of combined protein C and protein S deficiency in an individual.
Authorship and contributorship
TWS, ISRC and VCS designed and performed the molecular genetic experiments, TWS and CJC recruited the patient; RTAM wrote the manuscript; all authors edited the manuscript.
Funding &n2021 Copyright OAT. All rights reserv
This work was supported in part by a grant from the Canadian Institutes of Health Research to RTAM. ISRC was supported by a Graduate Fellowship from Canadian Blood Services.
Competing interest
The authors disclose no conflicts of interest
References
- Bafunno V, Margaglione M (2010) Genetic basis of thrombosis. Clinical and Chemical Laboratory Medicine 48: S41-51.
- Griffin JH, Zlokovic BV, Mosnier LO (2012) Protein C anticoagulant and cytoprotective pathways. Int J Hematol 95: 333-345. [Crossref]
- Foster DC, Yoshitake S, Davie EW (1985) The nucleotide sequence of the gene for human protein C. Proc Natl Acad Sci U S A 82: 4673-4677. [Crossref]
- Plutzky J, Hoskins JA, Long GL, Crabtree GR (1986) Evolution and organization of the human protein C gene. Proc Natl Acad Sci U S A 83: 546-550. [Crossref]
- Schmidel DK, Tatro AV, Phelps LG, Tomkzak JA, Long GL (1990) Organization of the human protein S genes. Biochemistry 29: 7845-7852. [Crossref]
- Ploos van Amstel HK, Reitsma PH, van der Logt CP, Bertina RM (1990) Intron-exon organization of the active human protein S gene PS alpha and its pseudogene PS beta: duplication and silencing during primate evolution. Biochemistry 29: 7853-7861.
- Edenbrandt CM, Lundwall A, Wydro R, Stenflo J (1990) Molecular analysis of the gene for vitamin K dependent protein S and its pseudogene. Cloning and partial gene organization. Biochemistry 29: 7861-7868. [Crossref]
- Reitsma PH, Poort SR, Bernardi F, Gandrille S, Long GL, et al. (1993) Protein C deficiency: A database of mutations. For the Protein C & S subcommittee of the scientific and standardization committee of the international society on thrombosis and haemostasis. Thrombosis & Haemostasis 69: 77-84. [Crossref]
- Nizzi FA, Jr Kaplan HS (1999) Protein C and S deficiency. Seminars in Thrombosis & Haemostasis 25: 265-272.
- Gandrille S, Borgel D, Sala N, Espinosa-Parrilla Y, Simmonds R, et al. (2000) Protein S deficiency: a database of mutations--summary of the first update. Thromb Haemost 84: 918. [Crossref]
- Lanke E, Johansson AM, Hillarp A, Lethagen S, Zöller B, et al. (2004) Co-segregation of the PROS1 locus and protein S deficiency in families having no detectable mutations in PROS1. J Thromb Haemost 2: 1918-1923. [Crossref]
- Kattel SBH, Regmi S, Neopane A (2011) Combined protein C and protein S deficiency presenting as deep vein thrombosis. Journal of Coagulation Disorders 3: 71-73.
- Formstone CJ, Hallam PJ, Tuddenham EGD, Voke J, Layton M, et al. (1996) Severe perinatal thrombosis in double and triple heterozygous offspring of a family segregating two independent protein S mutations and a protein C mutation. Blood 87: 3731-3737. [Crossref]
- Mondal R, Nandi M, Dhibar T (2010) Protein C and protein S deficiency presenting as deep venous thrombosis. Indian Pediatr 47: 188-189. [Crossref]
- Bishnu S, Regmi S, Neopane A, Karki DB (2011) Combined protein C and protein S deficiency presenting as deep vein thrombosis. Journal of Coagulation Disorders 3: 71-73.
- Sayin MR, Akpinar I, Karabag T, Aydin M, Dogan SM, et al., 2012. Left main coronary artery thrombus resulting from combined protein C and S deficiency. Intern Med 51: 3014-3044. [Crossref]
- Hayashida M, Yamada H, Yamazaki S, Nomura H, Yoshimura K, et al. (2003) Combined protein C and protein S deficiency in a family with repetitive thromboembolism and segregated gene mutations. Intern Med 42: 268-272. [Crossref]
- Patel ML, Sachan R, Seth G (2013) Combined deficiency of proteins C and S: ischaemic stroke in young individuals. BMJ Case Reports.
- Biguzzi E, Razzari C, Lane DA, Castaman G, Cappellari A, et al. (2005) Molecular diversity and thrombotic risk in protein S deficiency: the PROSIT study. Hum Mutat 25: 259-269. [Crossref]
- Zhang L, Castellino FJ (1993) The contributions of individual gamma-carboxyglutamic acid residues in the calcium-dependent binding of recombinant human protein C to acidic phospholipid vesicles. J Biol Chem 268: 12040-12045. [Crossref]
- Sanda N, Fujimori Y, Kashiwagi T, Takagi A, Murate T, et al. (2007) An Sp1 binding site mutation of the PROS1 promoter in a patient with protein S deficiency. Br J Haematol 138: 663-665. [Crossref]
- Tagariello G, Belvini D, Salviato R, Di Gaetano R, Zanotto D, et al. (2007) The Italian haemophilia B mutation database: a tool for genetic counselling, carrier detection and prenatal diagnosis. Blood Transfusion 5: 158-163. [Crossref]